Introduction
Amyloidosis is a systemic disease characterized by the extracellular deposition of insoluble proteins. In cardiac amyloidosis, or amyloid cardiomyopathy, amyloid fibrils accumulate in the interstitial spaces between myocytes, leading to cellular damage, reduced compliance, and increased stiffness. The condition is primarily caused by the deposition of 2 abnormally folded proteins: monoclonal immunoglobulin light chains and transthyretin, the latter also known as amyloid transthyretin (ATTR), a protein that transports thyroxine and retinol.[1] The most common form is amyloid light-chain amyloidosis, affecting more than 10 individuals per million annually. The 2nd form, ATTR amyloidosis, results from the deposition of either normal or mutated transthyretin proteins.[2] Other causes of amyloidosis are also discussed below (see Etiology).
Cardiac amyloidosis is the most common type of restrictive cardiomyopathy, with cardiac sarcoidosis and cardiac hemochromatosis as the other 2 variants. Infiltrative cardiomyopathies are characterized by depressed diastolic function in the presence of a nondilated left ventricle.[3] Regardless of etiology, cardiac amyloidosis is the leading cause of mortality in patients with systemic amyloidosis.[4]
Cardiac amyloidosis may present as a primary condition or be discovered incidentally in patients showing other signs and symptoms of systemic amyloidosis. Diagnostic and treatment delays are common.[5] Cardiac involvement in systemic amyloidosis has prognostic significance and is the most critical determinant of survival in these patients.[6]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Cardiac amyloidosis results from the extracellular deposition of a toxic substance known as amyloid—an aggregate of misfolded proteins combined with matrix-forming elements such as proteoglycans, glycosaminoglycans, collagen, and laminin. These misfolded proteins originate from 2 major sources: amyloid light chains and ATTR.
Microscopically, amyloid fibrils appear as nonbranching structures measuring 7 to 10 nm in diameter. The accumulation of these fibrils in the extracellular matrix causes myocardial stiffening and impaired cardiac performance. Diastolic dysfunction typically precedes systolic impairment, which emerges in the later stages of the disease.[7]
Cardiac amyloidosis has several etiological types. Primary amyloidosis, or amyloid light-chain amyloidosis, results from the deposition of light-chain fibrils produced by abnormal plasma cells in disorders such as multiple myeloma. Secondary amyloidosis, or amyloid A amyloidosis, is caused by the accumulation of serum amyloid A, an inflammatory protein elevated in chronic inflammatory diseases.
ATTR amyloidosis includes 2 forms. Wild-type ATTR (ATTRwt), previously known as senile systemic amyloidosis, arises from age-related deposition of normal transthyretin, while variant ATTR (ATTRv), formerly called "familial amyloidosis," results from over 130 known transthyretin mutations, the most common in the U.S. being a substitution of valine for isoleucine at position 122 (Val122Ile). Less common forms include hereditary apolipoprotein A-1 and A-4 amyloidosis, as well as isolated atrial amyloidosis, which involves amyloid derived from atrial natriuretic peptide.
Epidemiology
Cardiac amyloidosis is a rare but increasingly recognized disorder, with prevalence varying by etiology. Amyloid light-chain amyloidosis occurs in approximately 10% of patients with multiple myeloma, with cardiac involvement seen in 50% to 70% of those cases. The annual incidence of this condition is estimated at 1 per 100,000.
Familial transthyretin (ATTRv) amyloidosis is genetically heterogeneous, with over 100 transthyretin gene mutations identified. A study found the Val122Ile variant in 1.73% of a cohort of 14,333 African American individuals.[8]
Wild-type ATTR (ATTRwt) amyloidosis, formerly known as senile systemic amyloidosis, is the most common form of cardiac amyloidosis, present in over 10% of individuals older than 60 years and frequently misdiagnosed as heart failure with preserved ejection fraction. ATTRwt is also seen in at least 10% of patients with aortic stenosis and 10% to 15% of adults older than 65 with heart failure and preserved ejection fraction.[9] As survival and diagnostic accuracy improve, the prevalence of cardiac amyloidosis has risen, increasing from 8 to 17 per 100,000 person-years over the past 12 years.[10]
Pathophysiology
Amyloid deposition contributes to cardiac dysfunction through multiple mechanisms. Direct interstitial infiltration increases ventricular wall thickness and stiffness, leading to ventricular diastolic dysfunction. In amyloid light-chain amyloidosis, amyloid deposits may affect arterioles, resulting in angina or, rarely, myocardial infarction. Atrial infiltration promotes interstitial changes that provide a substrate for atrial fibrillation. Even in sinus rhythm, atrial amyloidosis elevates the risk of atrial thrombosis and thromboembolism. Amyloid light-chain amyloidosis also causes direct myocardial injury, as circulating light chains induce oxidative stress through reactive oxygen species.
The source of amyloid differs by type. In amyloid light-chain amyloidosis, fibrils are derived from abnormal light chains produced by plasma cells. In ATTR amyloidosis, the amyloid is formed from oligomers and monomers released due to destabilization of the transthyretin tetramer, either from aging or mutation. Isolated atrial amyloidosis, on the other hand, involves amyloid formed from the atrial natriuretic peptide.
Progressive myocardial infiltration disrupts normal contraction, leading to reduced stroke volume. Although the left ventricular ejection fraction may initially remain preserved, it declines as infiltration advances. Amyloid deposition in the sinoatrial node, atrioventricular node, and His-Purkinje system can result in various conduction blocks. In ATTR amyloidosis, especially ATTRv, amyloid infiltration of the autonomic nervous system may cause orthostatic hypotension, syncope, and gastrointestinal motility issues.
History and Physical
Patients with amyloidosis may either present with primary cardiac symptoms or be diagnosed incidentally during the evaluation for other systemic involvements. Symptoms of cardiac amyloidosis often overlap with those of heart failure and may include fatigue, dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, lower limb swelling, and abdominal distension. Additionally, patients may experience arrhythmia-related symptoms such as palpitations, chest pain, presyncope, or syncope.
Patients may present with musculoskeletal manifestations. Symptoms like tingling and numbness in the thumb, index, middle, and half of the ring finger, along with wrist pain or difficulty gripping objects, may indicate carpal tunnel syndrome. Some individuals may also report lower back pain related to lumbar stenosis. Polyneuropathy is another key symptom and can lead to muscle weakness, difficulty walking, and falls. Painful neuropathy in the hands and feet is common. Autonomic dysfunction is frequently seen and may present as chronic diarrhea, constipation, weight loss, erectile dysfunction, and orthostatic hypotension.
On physical examination, general findings may include periorbital edema, elevated jugular venous pressure, pedal edema, and macroglossia. Blood pressure may reveal postural hypotension. The precordial examination may show S3 or S4 heart sounds, which suggest heart failure or reduced compliance, respectively. Heart sounds may be muffled in cases of pericardial effusion. The abdominal exam might reveal ascites or hepatomegaly. A positive Popeye sign could suggest a distal or biceps tendon rupture. Additionally, signs of carpal tunnel syndrome or neuropathies may be observed during the musculoskeletal examination.[11]
Evaluation
The 2023 American College of Cardiology Expert Consensus on Cardiac Amyloidosis emphasizes the importance of following a diagnostic algorithm for patients with suspected cardiac amyloidosis. A high degree of clinical suspicion is required to make the diagnosis. Physicians, especially cardiologists, should be attentive to both cardiac and systemic manifestations of amyloidosis and begin the evaluation when clues suggest the presence of this disorder.
Clinical manifestations to consider include symptoms such as fatigue, heart failure, bilateral carpal tunnel syndrome, spontaneous tendon rupture, lumbar stenosis or pain, knee or hip replacement, peripheral neuropathy, autonomic dysfunction, and erectile dysfunction. Signs like intolerance to vasodilators, orthostatic hypotension, gastroparesis, urinary incontinence, and nephrotic syndrome should also be noted.
Initial evaluations may reveal features such as a pseudo-infarction pattern, atrial fibrillation, conduction abnormalities, or discordance between voltage on an electrocardiogram (ECG) and ventricular wall thickness seen on imaging. Other indicators include greater ventricular wall thickness, biatrial enlargement, progressive diastolic dysfunction, and apical sparing strain patterns on echocardiogram. Cardiac magnetic resonance (CMR) imaging may show diffuse subendocardial or transmural hyperenhancement. Persistently elevated low-level troponin I or elevated N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels may also raise suspicion.
Once cardiac amyloidosis is suspected, the first step is to exclude amyloid light-chain amyloidosis through a series of 3 monoclonal protein tests: serum κ/λ free light chain assay, serum immunofixation electrophoresis (SIFE), and urine immunofixation electrophoresis (UIFE). The serum κ/λ ratio is abnormal if it is less than 0.26 or greater than 1.65. If this ratio and the SIFE or UIFE results are normal, amyloid light-chain amyloidosis may be excluded with a negative predictive value of 99%.
Cardiac scintigraphy should be ordered when light chain assays are negative and ATTR amyloidosis is suspected. This evaluation may be performed simultaneously with the light chain assays, but the results should be interpreted together. Cardiac scintigraphy involves the administration of technetium pyrophosphate (Tc-PYP), and both planar and single photon emission computed tomography (SPECT) or single photon emission computed tomography combined with computed tomography (SPECT-CT) images should be acquired 1 or 3 hours after injection. A grade 2 or 3 uptake confirms the diagnosis of ATTR amyloidosis. Cardiac amyloidosis is unlikely if no uptake is observed. If the scintigraphy is positive, ATTR gene testing is recommended to distinguish between ATTRwt and ATTRv.
If monoclonal assays are positive, consultation with a hematologist for a biopsy is necessary. A positive Congo red stain or tissue typing via mass spectrometry can confirm the diagnosis of amyloid light-chain amyloidosis.
Cardiac amyloidosis remains underdiagnosed. The condition's varied systemic manifestations and symptoms, which differ based on etiology, often contribute to its underrecognition. However, patients with cardiac amyloidosis often present with specific findings on cardiovascular investigations, making early detection and diagnosis critical.
Electrocardiography
The 12-lead ECG may reveal a pseudo-infarction pattern, with low voltages in the limb leads and Q waves in the anterior and inferior leads (see Image. Cardiac Amyloidosis on Electrocardiography). The tracing may also show varying degrees of atrioventricular block, most commonly 1st-degree atrioventricular block. These findings are more typical of amyloid light-chain amyloidosis. In contrast, patients with ATTR amyloidosis are more likely to exhibit left bundle branch block, a higher degree of atrioventricular block, and generally normal limb-lead voltages with nonspecific ST-T segment changes. Atrial fibrillation is frequently observed in these patients.[12]
Echocardiography
Echocardiography often provides the first clue and plays a central role in both screening and diagnosing cardiac amyloidosis. A hallmark finding is increased myocardial wall thickness with a characteristic sparkling or granular appearance of the myocardium (see Image. Cardiac Amyloidosis on Transthoracic Echocardiogram).[13] Biatrial enlargement is almost always present. The left ventricle is typically nondilated, and systolic function tends to remain preserved until the later stages of the disease. Diastolic dysfunction, however, is universally observed and typically progresses rapidly in severity. Additional findings often include pericardial and pleural effusions.
Global longitudinal strain analysis on echocardiogram reveals a reduction in longitudinal strain from base to apex, with relative preservation of apical strain, producing the classic “cherry on top” appearance, which is pathognomonic for cardiac amyloidosis (see Image. Global Longitudinal Strain Analysis for Cardiac Amyloidosis). An apical-to-basal strain ratio greater than 1.1 is highly sensitive and specific for the condition.[14]
Clinical features such as syncope or presyncope, angina, heart failure symptoms, left ventricular hypertrophy with a hyperechoic (“bright”) myocardial texture, voltage-to-mass discordance on ECG, and the characteristic apical sparing pattern of strain imaging all strengthen the suspicion of cardiac amyloidosis.
Subtle but important differences exist between ATTR and amyloid light-chain amyloidosis on echocardiographic assessment. In amyloid light-chain amyloidosis, the left ventricle is generally symmetrically hypertrophied. In contrast, ATTR amyloidosis more often shows asymmetric hypertrophy, sometimes with a sigmoid septum configuration. Additionally, ATTR amyloidosis tends to cause a more significant decline in both systolic and diastolic left ventricular function, as well as greater increases in left and right ventricular masses, than amyloid light-chain amyloidosis.[15]
Cardiac Magnetic Resonance Imaging
CMR has become an essential tool in the diagnosis of cardiac amyloidosis. This modality can distinguish amyloidosis from other causes of increased myocardial thickness, such as hypertensive heart disease and cardiac sarcoidosis. Beyond structural assessment, CMR provides valuable insights into tissue characterization, allowing for the detection of early cardiac involvement. However, this tool cannot differentiate between ATTR and amyloid light-chain amyloidosis.
Characteristic CMR findings include restrictive ventricular morphology and diastolic dysfunction, often accompanied by disproportionate biatrial enlargement and increased myocardial mass and wall thickening. On late gadolinium enhancement (LGE), the affected segments show reduced systolic thickening. A hallmark feature is diffuse, circumferential subendocardial late gadolinium enhancement that does not conform to any single coronary artery territory. In more advanced disease, this enhancement may become transmural.
Native T1 values and extracellular volume fractions are elevated due to extracellular amyloid deposition.[16] The scan typically does not show myocardial edema, and gadolinium contrast rapidly washes out of the left ventricular blood pool. A notable and specific feature is the difficulty in appropriately nulling the normal myocardium on inversion time mapping. In healthy individuals, the left ventricular blood pool nulls before the myocardium. In cardiac amyloidosis, this pattern is reversed—myocardial nulling occurs prior to blood pool nulling, which is considered a highly suggestive finding.[17]
Nuclear Single Photon Emission Computed Tomography
A strongly positive bone tracer cardiac scintigraphy—defined as cardiac uptake greater than bone (grade 2 or 3) or a heart-to-chest wall uptake ratio of more than 1.5 at 1 hour—combined with echocardiographic features suggestive of cardiac amyloidosis and the absence of systemic signs or symptoms of multiple myeloma, is considered nearly diagnostic of ATTR amyloidosis. In contrast, patients with amyloid light-chain amyloidosis typically demonstrate little to no cardiac uptake of the radiotracer.[18]
A semiquantitative visual grading system is applied when using technetium-labeled phosphate derivatives to diagnose ATTR cardiac amyloidosis. Radiotracer uptake is compared to rib bone activity and graded as follows:
- Grade 0: No uptake (normal)
- Grade 1: Less than bone uptake (mild)
- Grade 2: Uptake equal to bone (moderate)
- Grade 3: Uptake greater than bone (severe)
A grade 2 or 3 uptake in the absence of monoclonal proteins confirms a diagnosis of ATTR cardiac amyloidosis.
Endomyocardial Biopsy
Endomyocardial biopsy remains the gold standard for diagnosing cardiac amyloidosis. This assessment reveals the presence of amyloid fibrils, which appear salmon-colored with Congo red staining. Under polarized light microscopy, these fibrils display the characteristic apple-green birefringence. Although invasive, cardiac biopsy is highly sensitive, with a reported sensitivity ranging from 87% to 98%.[19]
Genotyping
Genetic testing is crucial in patients with ATTR amyloidosis. To date, over 120 ATTR gene variants have been linked to cardiac amyloidosis. Among these, threonine 60 to alanine (Thr60Ala) and valine 122 to isoleucine (Val122Ile) are the most common. Genotyping is essential for predicting treatment response and clinical prognosis. Variant prevalence varies by geographic region and ethnicity.[20]
Treatment / Management
The treatment of cardiac amyloidosis is multifaceted, reflecting the complex interplay between heart failure, arrhythmias, and the underlying amyloid pathology. An individualized approach that integrates volume management, disease-modifying therapies, and rhythm control is critical to improving both quality of life and survival.
Heart Failure and Volume Management
Volume management is central to the treatment of cardiac amyloidosis. In patients with amyloid light-chain amyloidosis, diuretics remain the cornerstone of therapy. However, these patients often have poor tolerance for renin-angiotensin-aldosterone system inhibitors, as even low doses can precipitate significant hypotension. In contrast, patients with ATTR amyloidosis generally tolerate these medications better.
Orthostatic hypotension is frequently encountered due to autonomic dysfunction. In such cases, peripheral vasoconstrictors like midodrine may be used to support blood pressure while allowing continued diuresis to relieve congestion.
β-blockers are also poorly tolerated. The development of profound hypotension after initiating even low doses should raise clinical suspicion for underlying cardiac amyloidosis. Consequently, mineralocorticoid receptor antagonists and loop diuretics remain essential components of heart failure management in these patients.
Sodium-glucose cotransporter 2 (SGLT-2) inhibitors may offer benefits in selected patients, although data specific to cardiac amyloidosis remains limited. Thiazide diuretics, such as metolazone, should be used cautiously due to the risk of excessive diuresis, hypokalemia, and renal dysfunction.
Heart transplantation may be considered in carefully selected patients. In amyloid light-chain amyloidosis, orthotopic heart transplantation is associated with a significant risk of disease recurrence in the transplanted heart. However, for patients with clinically isolated cardiac involvement who are willing to undergo intensive chemotherapy after transplant, this strategy may be viable. The rationale is that the amyloid-affected heart cannot tolerate aggressive chemotherapy due to reduced myocardial reserve. Therefore, a transplant followed by chemotherapy can improve outcomes, with an expected 5-year survival rate of approximately 60%.
Patients with ATTRwt amyloidosis typically have isolated cardiac involvement, making them potential candidates for heart transplantation. Nonetheless, advanced age at diagnosis—usually in the 7th decade or beyond—often precludes them from being eligible transplant recipients. For individuals with end-stage heart failure who are not transplant candidates, early involvement of palliative care services is essential to ensure appropriate symptom management and quality-of-life support.
Disease-Modifying Therapy
Tafamidis is currently the only oral medication approved by the U.S. Food and Drug Administration (FDA) for the treatment of ATTR amyloid cardiomyopathy. This agent is indicated for both ATTRv and ATTRwt forms. Tafamidis functions by stabilizing the tetrameric structure of the transthyretin protein, thereby preventing its dissociation into monomers—a critical early event in the pathogenesis of amyloid fibril formation.
Findings from the Transthyretin Amyloidosis Cardiomyopathy Clinical Trial (ATTR-ACT) demonstrated that tafamidis significantly reduced all-cause mortality and cardiovascular-related hospitalizations over a 30-month period when compared to placebo. These benefits were consistent across key subgroups, including both hereditary and wild-type disease, and across New York Heart Association (NYHA) functional classes I through III.[21] A post hoc analysis further revealed that tafamidis attenuated the progressive decline in both systolic and diastolic left ventricular function, suggesting a cardioprotective effect that may slow disease progression.[22](A1)
The FDA-approved doses for ATTR amyloid cardiomyopathy are tafamidis 61 mg and tafamidis meglumine 80 mg. Despite its demonstrated clinical efficacy, access to this therapy is often limited by its high cost, which remains a primary barrier to widespread use.
For patients with ATTRv accompanied by polyneuropathy, FDA-approved transthyretin silencers, such as Patisiran, Inotersen, and Vutrisiran, are available.[23] These drugs function by targeting ATTR mRNA, thereby reducing hepatic production of the mutant protein and, consequently, limiting further amyloid deposition. Management in such cases typically involves interprofessional collaboration with neurology specialists.
In the context of amyloid light-chain cardiac amyloidosis, treatment necessitates coordinated management between hematologists and cardiologists. The principal therapeutic objective is the elimination of circulating monoclonal light chains and the suppression of clonal plasma cell activity within the bone marrow. Commonly employed chemotherapeutic agents include melphalan, an alkylating agent, and bortezomib, a proteasome inhibitor. Bortezomib is frequently combined with dexamethasone and cyclophosphamide to enhance therapeutic efficacy.
A favorable cardiac response is typically defined by at least a 30% reduction in B-type natriuretic peptide levels over a 6-month interval. For patients eligible for autologous stem cell transplantation (SCT), high-dose melphalan followed by transplantation is considered the standard of care. In patients who are unsuitable for transplantation, the preferred regimen is Dara-CyBorD therapy, which includes daratumumab, cyclophosphamide, bortezomib, and dexamethasone.
Management of Arrhythmias
Depending on the type of arrhythmia, patients with cardiac amyloidosis may require rate or rhythm control in addition to anticoagulation, particularly for atrial fibrillation. Interventions may include a permanent pacemaker for atrioventricular block or an implantable cardioverter-defibrillator for ventricular tachycardia or aborted sudden cardiac death.
Rhythm and rate control in atrial arrhythmias is particularly challenging, as β-blockers are often poorly tolerated due to their hypotensive effects. Profound hypotension with even low doses should prompt consideration of underlying cardiac amyloidosis. Amiodarone is a reasonable and generally well-tolerated alternative in this population.
Digoxin tends to bind to amyloid fibrils, increasing the risk of toxicity. However, with careful dosing and monitoring, this medication may still be used for rate control in patients with atrial fibrillation. Calcium channel blockers have not been shown to provide significant benefit in the setting of amyloid-related diastolic dysfunction, such as that seen in hypertensive heart disease, and may exacerbate hypotension.
Catheter ablation may be attempted for atrial flutter, although this intervention carries a high recurrence rate in amyloidosis-related atrial fibrillation. Anticoagulation is often necessary regardless of rhythm or symptom burden due to the elevated risk of intracardiac thrombus formation and thromboembolism.
In patients with atrioventricular block, biventricular pacing is preferred over right ventricular pacing alone. Right ventricular pacing in the context of a stiff, amyloid-infiltrated myocardium may worsen cardiac performance. Implantable cardioverter-defibrillators for primary prevention are generally not recommended, as they lack proven survival benefits. The use of these devices should be reserved for secondary prevention in patients with a documented life-threatening ventricular arrhythmia.
Liver Transplantation
Liver transplantation plays a role in the treatment of ATTRv but is not typically performed for ATTRwt or amyloid light-chain amyloidosis. The benefit of liver transplantation in ATTRv arises from the fact that transthyretin is primarily produced in the liver. Replacing the liver halts the production of mutated transthyretin, which can prevent further amyloid deposition.
Liver transplantation is traditionally considered for ATTRv patients who are at risk of neuropathy, as a heart transplant alone may exacerbate or accelerate the progression of neuropathy. In the era of transthyretin silencers, the criteria for heart-liver transplantation are not well established, and ongoing research is needed to define its role in amyloidosis treatment.
Palliative Care
Patients with cardiac amyloidosis often require palliative care, with specific interventions varying based on disease type and stage. Measures involve establishing timely treatment goals, discussing patient preferences, and managing symptoms early to help patients live as actively as possible. Pain relief, psychological and spiritual support, and affirming life and death as natural processes are essential components. Additionally, providing family support is crucial throughout the palliative care process.
Differential Diagnosis
Any form of restrictive cardiomyopathy can be mistaken for cardiac amyloidosis. Shared features include dyspnea despite a preserved ejection fraction, diastolic dysfunction, and biatrial enlargement. Differential diagnoses encompass conditions such as cardiac sarcoidosis, glycogen storage disorders, and hemochromatosis. Other mimics with similar echocardiographic findings include hypertensive heart disease and hypertrophic cardiomyopathy.
Prognosis
The prognosis of cardiac amyloidosis is variable and depends on the specific type. Median survival for untreated patients is approximately 6 to 12 months for amyloid light-chain amyloidosis, 3.6 to 4.8 years for ATTRwt cardiac amyloidosis, 2.6 years for ATTRV cardiac amyloidosis due to the Val122Ile mutation, and 5.8 years for ATTRv cardiac amyloidosis caused by other transthyretin mutations. Overall, ATTR amyloidosis has a better prognosis than amyloid light-chain amyloidosis. ATTR amyloidosis progresses slowly and typically presents later in life, most commonly during the 7th decade.
In patients with amyloid light-chain amyloidosis who undergo SCT, the 4-year survival rate exceeds 90%. For those with cardiac involvement who receive a transplant, median survival can extend beyond 10 years. In general, amyloid light-chain amyloidosis carries a median survival of about 10 years, except in advanced-stage disease, where the 1-year survival drops to 50%.
Mutant ATTR amyloidosis has a 4-year survival of approximately 16%. The prognosis depends heavily on the specific genetic variant. The Val30Met mutation—the most common—has an overall survival rate of 79%, whereas the Val122Ile mutation is associated with a less favorable 4-year prognosis of 40%.[24]
Markers of poor prognosis include elevated troponin I and NT-proBNP, left ventricular wall thickness greater than 15 mm, advanced diastolic dysfunction, multisystem involvement, and the presence of arrhythmias. The Mayo Clinic staging system is a standard tool used to stratify prognosis in cardiac amyloidosis, and it employs biomarkers to assess disease severity and predict outcomes.
Amyloid light-chain amyloidosis staging is determined based on cardiac biomarker thresholds. A worse prognosis is associated with an NT-proBNP level over 1,800 pg/mL, a troponin T level greater than 0.025 ng/mL, and a serum-free light chain difference above 18 mg/dL. Stage I is defined by all 3 values being below these cutoffs and is associated with a median survival of approximately 5 years. Stage II is assigned when 1 of the 3 biomarkers exceeds its threshold, with a median survival of around 3.5 years. Stage III is defined by 2 elevated markers and carries a median survival of about 1.5 years. Stage IV, in which all 3 markers are elevated, is associated with a median survival of roughly 6 months.
ATTR amyloidosis staging relies on NT-proBNP and troponin T levels. Stage I is defined by an NT-proBNP of 3,000 pg/mL or less and a troponin T of 0.05 ng/mL or less, with a median survival of approximately 5.7 years. Stage II includes patients who exceed either of those thresholds and is associated with a median survival of around 4.2 years. Stage III applies when both NT-proBNP is above 3,000 pg/mL and troponin T is greater than 0.05 ng/mL, with a median survival of about 2.5 years.[25]
Complications
Complications of cardiac amyloidosis primarily arise from structural abnormalities in the heart, which significantly impact a patient's health. Atrial fibrillation is a common sequela, which can result in thromboembolism and heart failure, further worsening the clinical course. Diastolic dysfunction, often seen in cardiac amyloidosis, contributes to heart failure, increases mortality, and promotes arrhythmogenesis. Additionally, frequent hospitalizations due to heart failure and an increased risk of mortality are common challenges. Ventricular arrhythmias and cardiac conduction abnormalities are other serious concerns contributing to an increased risk of sudden cardiac events. Systemic involvement in amyloidosis can also lead to autonomic neuropathy. Tendinopathies may also arise in systemic involvement, affecting the patient's mobility and quality of life.
Deterrence and Patient Education
Cardiac amyloidosis has a variable prognosis, with treatment modalities differing based on the underlying etiology. Patients must understand the course of the disease, as well as its systemic nature. Patients should also recognize the need for an interprofessional team to manage their care, including specialists in cardiology, hematology, gastroenterology, neurology, and transplantation medicine.
Heart failure management in cardiac amyloidosis differs from standard guideline-based therapy. Patients often have heightened sensitivity to even the smallest doses of medication, requiring a very gradual titration. Thus, more frequent follow-up visits are necessary compared to heart failure due to non-amyloid-related causes.
Several factors limit access to care, including the cost and availability of novel therapeutics. Barriers to equitable care include restricted access to specialist care, variations in out-of-pocket costs, the high expense of medications, and the complexity of insurance approval processes.
Enhancing Healthcare Team Outcomes
The prognosis of cardiac amyloidosis heavily depends on early diagnosis, which underscores the need for an interprofessional approach to management. Clinicians, including general practitioners, internists, and clinical cardiologists, should maintain a high degree of suspicion based on the clinical history and laboratory investigations, which can lead to earlier diagnosis and quicker initiation of therapy.
Managing amyloid light-chain amyloidosis requires collaboration between cardiologists and hematologists to assess candidacy for SCT, ensure cardiac status is stable for SCT, monitor cardiotoxicity of therapy, and assess candidacy for a heart transplant. Managing ATTRv cardiac amyloidosis with cardiomyopathy and neuropathy requires coordination between cardiologists and neurologists for initiating and monitoring disease-modifying therapies.
A gastrointestinal specialist is required for the management of gastrointestinal amyloidosis. Palliative care collaboration is extremely crucial, as many patients eventually require comprehensive support. Nurse practitioners and rehabilitation specialists are an integral part of the palliative care team.
Media
(Click Image to Enlarge)
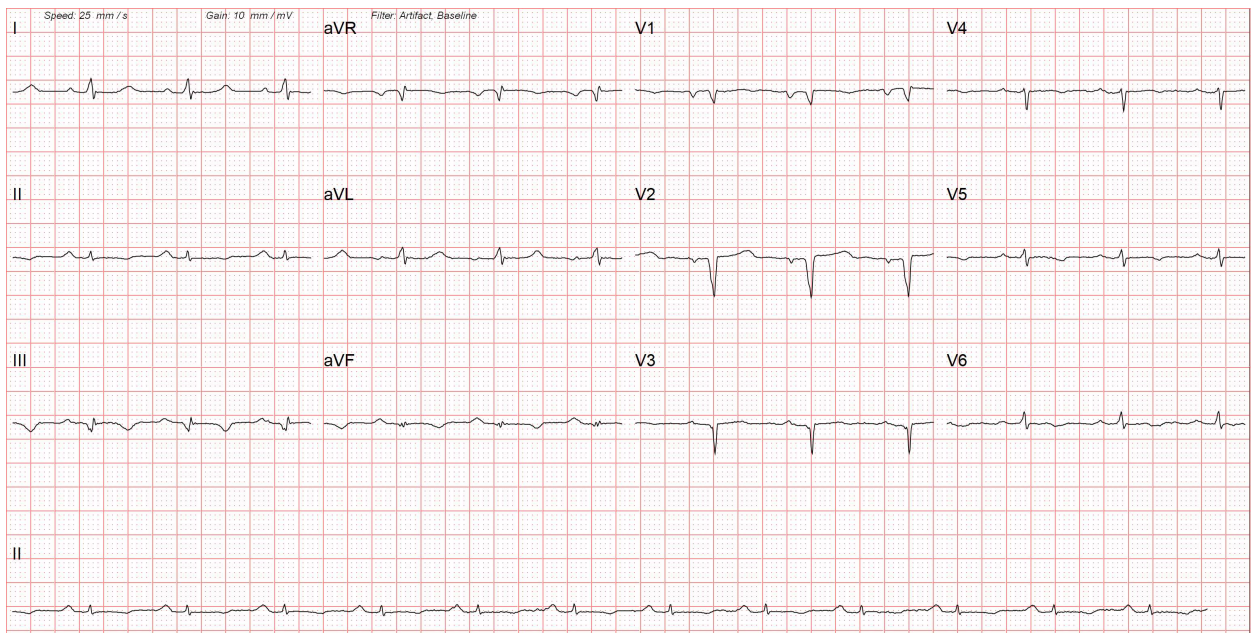
Cardiac Amyloidosis on Electrocardiography. This image shows low-voltage complexes in the limb leads due to diffuse myocardial infiltration. The 12-lead electrocardiogram also reveals Q waves in the anterior chest leads, indicative of myocardial fibrosis, which results in a pseudo-infarction pattern.
Contributed by Pirbhat Shams, MBBS
(Click Image to Enlarge)
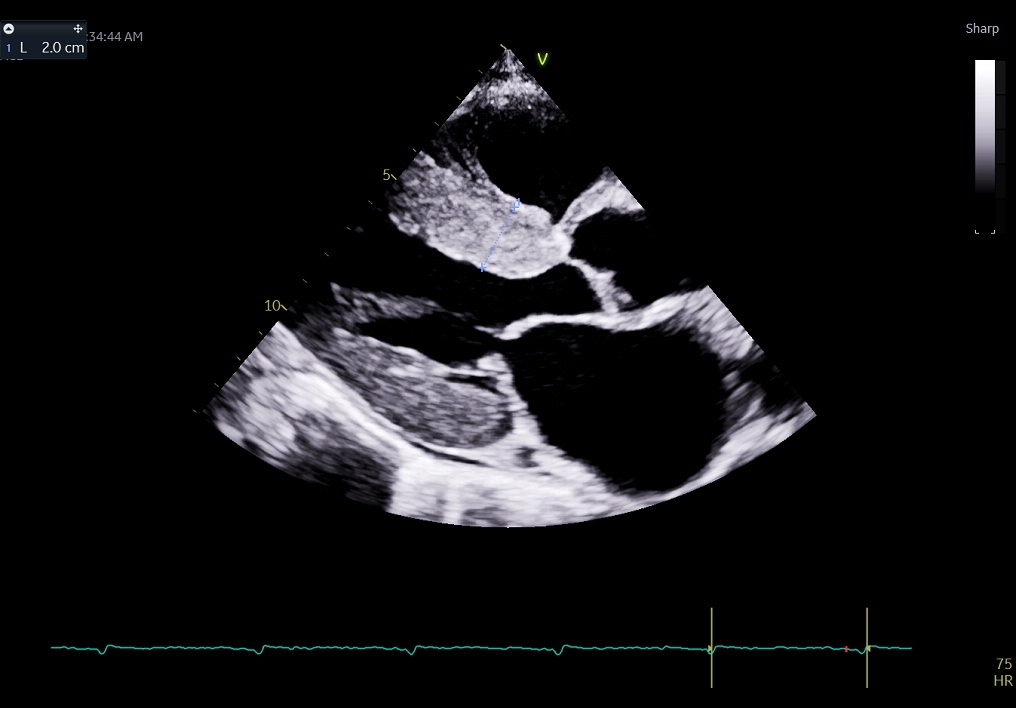
Cardiac Amyloidosis on Transthoracic Echocardiogram. This parasternal long-axis view shows a thickened, bright myocardium, often the first clue in diagnosing cardiac amyloidosis. Despite the thickened myocardium, the patient exhibited a low-voltage ECG (bottom).
Contributed by Pirbhat Shams, MBBS
(Click Image to Enlarge)
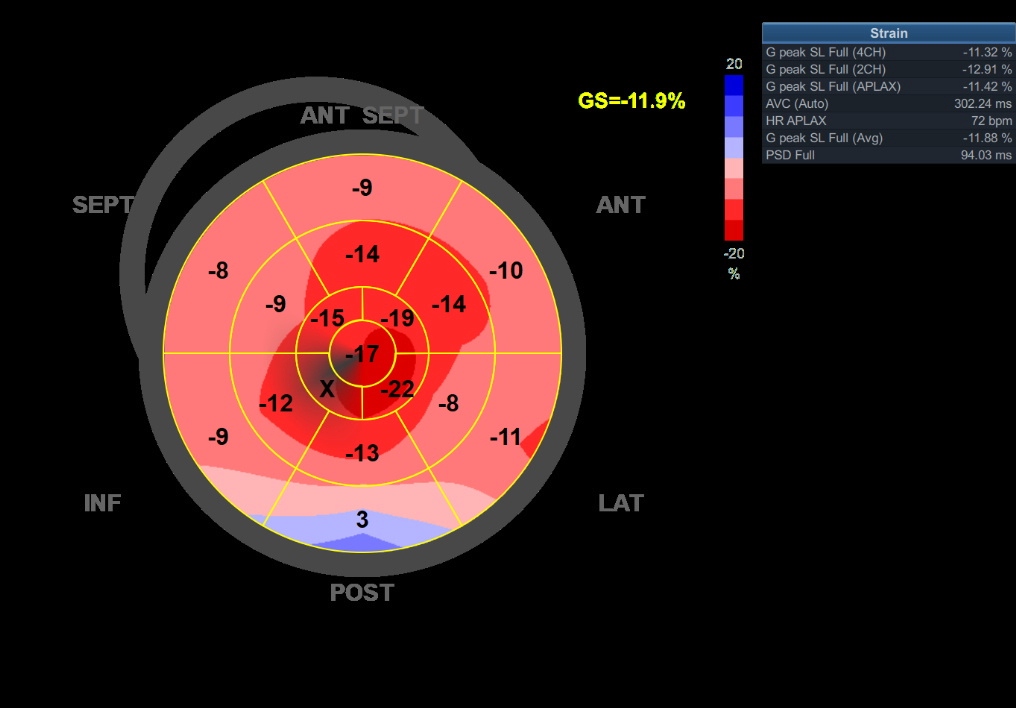
Global Longitudinal Strain Analysis for Cardiac Amyloidosis. This image shows the global longitudinal strain of a patient with cardiac amyloidosis, highlighting the typical "cherry red spot" on a bull's-eye map. The cherry-red spot indicates reduced base-to-mid strain, with relative sparing of the left ventricular apex.
Contributed by Pirbhat Shams, MBBS
References
Writing Committee, Kittleson MM, Ruberg FL, Ambardekar AV, Brannagan TH, Cheng RK, Clarke JO, Dember LM, Frantz JG, Hershberger RE, Maurer MS, Nativi-Nicolau J, Sanchorawala V, Sheikh FH. 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee. Journal of the American College of Cardiology. 2023 Mar 21:81(11):1076-1126. doi: 10.1016/j.jacc.2022.11.022. Epub 2023 Jan 23 [PubMed PMID: 36697326]
Level 3 (low-level) evidenceNienhuis HL, Bijzet J, Hazenberg BP. The Prevalence and Management of Systemic Amyloidosis in Western Countries. Kidney diseases (Basel, Switzerland). 2016 Apr:2(1):10-9. doi: 10.1159/000444206. Epub 2016 Feb 25 [PubMed PMID: 27536687]
Brown KN, Pendela VS, Ahmed I, Diaz RR. Restrictive Cardiomyopathy. StatPearls. 2025 Jan:(): [PubMed PMID: 30725919]
Martinez-Naharro A, Hawkins PN, Fontana M. Cardiac amyloidosis. Clinical medicine (London, England). 2018 Apr 1:18(Suppl 2):s30-s35. doi: 10.7861/clinmedicine.18-2-s30. Epub [PubMed PMID: 29700090]
Ladefoged B, Dybro A, Povlsen JA, Vase H, Clemmensen TS, Poulsen SH. Diagnostic delay in wild type transthyretin cardiac amyloidosis - A clinical challenge. International journal of cardiology. 2020 Apr 1:304():138-143. doi: 10.1016/j.ijcard.2019.12.063. Epub 2020 Jan 25 [PubMed PMID: 32033783]
Lei C, Zhu X, Hsi DH, Wang J, Zuo L, Ta S, Yang Q, Xu L, Zhao X, Wang Y, Sun S, Liu L. Predictors of cardiac involvement and survival in patients with primary systemic light-chain amyloidosis: roles of the clinical, chemical, and 3-D speckle tracking echocardiography parameters. BMC cardiovascular disorders. 2021 Jan 21:21(1):43. doi: 10.1186/s12872-021-01856-3. Epub 2021 Jan 21 [PubMed PMID: 33478398]
Siddiqi OK, Ruberg FL. Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment. Trends in cardiovascular medicine. 2018 Jan:28(1):10-21. doi: 10.1016/j.tcm.2017.07.004. Epub 2017 Jul 13 [PubMed PMID: 28739313]
Level 2 (mid-level) evidenceJacobson DR, Alexander AA, Tagoe C, Buxbaum JN. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis. 2015:22(3):171-4. doi: 10.3109/13506129.2015.1051219. Epub 2015 Jul 2 [PubMed PMID: 26123279]
Bajwa F, O'Connor R, Ananthasubramaniam K. Epidemiology and clinical manifestations of cardiac amyloidosis. Heart failure reviews. 2022 Sep:27(5):1471-1484. doi: 10.1007/s10741-021-10162-1. Epub 2021 Oct 25 [PubMed PMID: 34694575]
Gilstrap LG, Dominici F, Wang Y, El-Sady MS, Singh A, Di Carli MF, Falk RH, Dorbala S. Epidemiology of Cardiac Amyloidosis-Associated Heart Failure Hospitalizations Among Fee-for-Service Medicare Beneficiaries in the United States. Circulation. Heart failure. 2019 Jun:12(6):e005407. doi: 10.1161/CIRCHEARTFAILURE.118.005407. Epub 2019 Jun 7 [PubMed PMID: 31170802]
Porcari A, Rossi M, Cappelli F, Canepa M, Musumeci B, Cipriani A, Tini G, Barbati G, Varrà GG, Morelli C, Fumagalli C, Zampieri M, Argirò A, Vianello PF, Sessarego E, Russo D, Sinigiani G, De Michieli L, Di Bella G, Autore C, Perfetto F, Rapezzi C, Sinagra G, Merlo M. Incidence and risk factors for pacemaker implantation in light-chain and transthyretin cardiac amyloidosis. European journal of heart failure. 2022 Jul:24(7):1227-1236. doi: 10.1002/ejhf.2533. Epub 2022 May 16 [PubMed PMID: 35509181]
Cheng Z, Zhu K, Tian Z, Zhao D, Cui Q, Fang Q. The findings of electrocardiography in patients with cardiac amyloidosis. Annals of noninvasive electrocardiology : the official journal of the International Society for Holter and Noninvasive Electrocardiology, Inc. 2013 Mar:18(2):157-62. doi: 10.1111/anec.12018. Epub 2012 Nov 22 [PubMed PMID: 23530486]
Kiotsekoglou A, Saha SK, Nanda NC, Lindqvist P. Echocardiographic diagnosis of cardiac amyloidosis: Does the masquerader require only a "cherry on top"? Echocardiography (Mount Kisco, N.Y.). 2020 Nov:37(11):1713-1715. doi: 10.1111/echo.14952. Epub [PubMed PMID: 33283347]
Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation. 2017 Apr 4:135(14):1357-1377. doi: 10.1161/CIRCULATIONAHA.116.024438. Epub [PubMed PMID: 28373528]
Boldrini M, Cappelli F, Chacko L, Restrepo-Cordoba MA, Lopez-Sainz A, Giannoni A, Aimo A, Baggiano A, Martinez-Naharro A, Whelan C, Quarta C, Passino C, Castiglione V, Chubuchnyi V, Spini V, Taddei C, Vergaro G, Petrie A, Ruiz-Guerrero L, Moñivas V, Mingo-Santos S, Mirelis JG, Dominguez F, Gonzalez-Lopez E, Perlini S, Pontone G, Gillmore J, Hawkins PN, Garcia-Pavia P, Emdin M, Fontana M. Multiparametric Echocardiography Scores for the Diagnosis of Cardiac Amyloidosis. JACC. Cardiovascular imaging. 2020 Apr:13(4):909-920. doi: 10.1016/j.jcmg.2019.10.011. Epub 2019 Dec 18 [PubMed PMID: 31864973]
Kwong RY, Falk RH. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005 Jan 18:111(2):122-4 [PubMed PMID: 15657385]
Tipoo Sultan FA, Khan MT. Clinical presentation, diagnostic features on cardiac magnetic resonance imaging and outcome of patients with cardiac amyloidosis presenting to a tertiary care setting. JPMA. The Journal of the Pakistan Medical Association. 2021 Dec:71(12):2802-2805. doi: 10.47391/JPMA.385. Epub [PubMed PMID: 35150542]
Li W, Uppal D, Wang YC, Xu X, Kokkinidis DG, Travin MI, Tauras JM. Nuclear Imaging for the Diagnosis of Cardiac Amyloidosis in 2021. Diagnostics (Basel, Switzerland). 2021 May 30:11(6):. doi: 10.3390/diagnostics11060996. Epub 2021 May 30 [PubMed PMID: 34070853]
Bowen K, Shah N, Lewin M. AL-Amyloidosis Presenting with Negative Congo Red Staining in the Setting of High Clinical Suspicion: A Case Report. Case reports in nephrology. 2012:2012():593460. doi: 10.1155/2012/593460. Epub 2012 Dec 9 [PubMed PMID: 24555137]
Level 3 (low-level) evidenceMaurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, Steidley DE, Ventura H, Murali S, Silver MA, Jacoby D, Fedson S, Hummel SL, Kristen AV, Damy T, Planté-Bordeneuve V, Coelho T, Mundayat R, Suhr OB, Waddington Cruz M, Rapezzi C, THAOS Investigators. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). Journal of the American College of Cardiology. 2016 Jul 12:68(2):161-72. doi: 10.1016/j.jacc.2016.03.596. Epub [PubMed PMID: 27386769]
Level 3 (low-level) evidenceDamy T, Garcia-Pavia P, Hanna M, Judge DP, Merlini G, Gundapaneni B, Patterson TA, Riley S, Schwartz JH, Sultan MB, Witteles R. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. European journal of heart failure. 2021 Feb:23(2):277-285. doi: 10.1002/ejhf.2027. Epub 2020 Nov 12 [PubMed PMID: 33070419]
Shah SJ, Fine N, Garcia-Pavia P, Klein AL, Fernandes F, Weissman NJ, Maurer MS, Boman K, Gundapaneni B, Sultan MB, Elliott P. Effect of Tafamidis on Cardiac Function in Patients With Transthyretin Amyloid Cardiomyopathy: A Post Hoc Analysis of the ATTR-ACT Randomized Clinical Trial. JAMA cardiology. 2024 Jan 1:9(1):25-34. doi: 10.1001/jamacardio.2023.4147. Epub [PubMed PMID: 37966817]
Level 1 (high-level) evidenceKittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, Nair AP, Nativi-Nicolau J, Ruberg FL, American Heart Association Heart Failure and Transplantation Committee of the Council on Clinical Cardiology. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation. 2020 Jul 7:142(1):e7-e22. doi: 10.1161/CIR.0000000000000792. Epub 2020 Jun 1 [PubMed PMID: 32476490]
Aimo A, Merlo M, Porcari A, Georgiopoulos G, Pagura L, Vergaro G, Sinagra G, Emdin M, Rapezzi C. Redefining the epidemiology of cardiac amyloidosis. A systematic review and meta-analysis of screening studies. European journal of heart failure. 2022 Dec:24(12):2342-2351. doi: 10.1002/ejhf.2532. Epub 2022 May 16 [PubMed PMID: 35509173]
Level 1 (high-level) evidenceKumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, Laumann K, Zeldenrust SR, Leung N, Dingli D, Greipp PR, Lust JA, Russell SJ, Kyle RA, Rajkumar SV, Gertz MA. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012 Mar 20:30(9):989-95. doi: 10.1200/JCO.2011.38.5724. Epub 2012 Feb 13 [PubMed PMID: 22331953]