Introduction
Corticobasal ganglionic degeneration (CBD) is a rare and progressive neurodegenerative disorder with diverse clinical and pathological features. The most common clinical presentation, corticobasal syndrome, includes asymmetric limb rigidity, Parkinsonism, dystonia, and cortical dysfunction, such as apraxia or sensory deficits. Pathologically, CBD is associated with 4-repeat (4R; this refers to the presence of 4 microtubule-binding domains in the tau protein isoform) hyperphosphorylated tau protein inclusions in neurons and glial cells, forming astrocytic plaques. Despite advancements in understanding the pathology, the mechanisms underlying tau-induced neurodegeneration remain unclear, and disease-modifying treatments are unavailable. There is a need to delineate the disease's intricate pathophysiologic processes further.[1][2][3][4] Accurate early diagnosis is essential, as it can guide patient care and facilitate the development of targeted therapies and disease-specific biomarkers.
Interprofessional care plays a critical role in managing CBD, particularly given its progressive nature and the lack of curative treatments. Collaborative care teams—neurologists, physical therapists, occupational therapists, and palliative care specialists—can optimize symptom management and address functional impairments. Early discussions of end-of-life care and support for patients and families are essential in improving the quality of life for those with this debilitating condition.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
CBD is primarily considered an idiopathic or sporadic neurodegenerative disorder, with familial cases being exceedingly rare.[2][5] Given the diagnostic difficulty, little is known about the disorder’s risk factors. The only well-established risk factor for CBD is advanced age.[2] No evidence links environmental exposures to toxic or infectious agents in its pathogenesis. At its core, CBD is driven by the pathological accumulation of hyperphosphorylated 4R tau protein, which disrupts microtubule stability, leading to neurodegeneration. Affected regions include the cerebral cortex and basal ganglia, correlating with the disease's hallmark motor and cognitive symptoms. This tau accumulation results in characteristic pathological findings such as astrocytic plaques, oligodendrocytic coiled bodies, and neurofibrillary tangles.
Genetic Basis
Mutations in the microtubule-associated protein tau (MAPT) gene are central to the pathogenesis of CBD due to their role in disrupting membrane-associated 4R tau function.[6] The extended MAPT tau haplotype (H1) and homozygosity for H1/H1 significantly increase in pathologically confirmed CBD cases, similar to progressive supranuclear palsy (PSP).[7] The MAPT H1c allele, a regulatory region likely enhancing 4R tau transcript production, is particularly prevalent in CBD and PSP.[8][9] In contrast, the H2 haplotype appears protective against CBD.[10] Results from genome-wide association studies have reinforced the associations at the MAPT locus between CBD and PSP.[8] Certain MAPT mutations—such as those in exon 10, intron 10, and exon 13—can mimic the clinical and pathological features of corticobasal syndrome and CBD.[11][12]
A genome-wide association study involving pathologically confirmed CBD also identified associations at single nucleotide polymorphisms in non-MAPT loci, including chromosome 17q21 (MAPT expression), chromosome 8p12 (lnc-KIF13B-1, a long noncoding ribonucleic acid), and chromosome 2p22 (SOS1). Notably, the study's results revealed a shared genetic risk at 3p22 involving myelin-associated oligodendrocyte basic protein (MOBP), highlighting its potential pathogenic significance given the oligodendrocyte and white matter involvement in tauopathies like CBD and PSP.[8] Further research confirmed overlapping genetic risk factors between CBD and PSP at other non-MAPT loci, including MOBP, NSF (linked to the H1 haplotype), epidermal growth factor receptor, glycine dehydrogenase, and CXC motif chemokine receptor 4. Additionally, the genetic overlap between CBD and frontotemporal dementia appears predominantly localized to the MAPT locus.[13]
Evidence of broader genetic pleiotropy has also emerged, linking CBD to immune-mediated diseases such as celiac disease, suggesting immune dysfunction may play a role in its pathogenesis.[14] Exome sequencing of 2 familial CBD cases identified mutations in Mg²+ transporter (MRS2) and zinc-finger and homeodomain protein 2 (ZHX2), both regulated by MIR4277.[15] These findings underscore the complexity of CBD genetics and emphasize the need for further studies to clarify the roles of these genes in the disease's pathophysiology.
Epidemiology
CBD is a neurodegenerative disorder characterized by complex pathological mechanisms and diverse clinical presentations, collectively referred to as clinicopathologic heterogeneity. This variability significantly hampers antemortem diagnosis and limits the ability to conduct robust epidemiological studies. Notably, CBD pathology is correctly predicted in only 25% to 56% of cases prior to autopsy.[16]
Due to its diagnostic challenges, studies often focus on the broader syndrome of CBS when assessing incidence and prevalence. Results from a meta-analysis showed the incidence of CBS ranges from 0.03 to 0.8 per 100,000 person-years and its prevalence from 0.83 to 25 per 100,000. Data were drawn from North America, Europe, Singapore, Japan, Sydney, Buenos Aires, and Egypt.[17] Additional estimates suggest an annual CBS incidence of less than 1 per 100,000 patients based on study results from Japan and Russia.[18][19] While earlier reports indicated an annual CBD incidence of 0.6 to 0.9 per 100,000, representing 4% to 6% of Parkinsonian syndromes, and a prevalence of 4.9 to 7.3 per 100,000, results from a large United Kingdom cross-sectional study involving 121,608 patients did not identify any confirmed cases of CBD.[20][21]
The disease typically has a late onset, with the youngest pathologically confirmed case reported at 42 and an average age of onset around 65.[22][23] An autopsy series from the CurePSP Brain Bank at the Mayo Clinic (Jacksonville, Florida) examined 76 cases of pathologically confirmed CBD. They found a mean age of death of 70 ± 8.7 years (range 46–89 years), with 53% of the cases being men. The estimated average disease duration was 6 ± 2.3 years.[2] While results from some studies have suggested a higher prevalence of CBD in women, others have not demonstrated significant sex differences.[22][24][25]
Pathophysiology
The pathophysiology of CBD is primarily driven by the accumulation of 4R hyperphosphorylated tau protein, which aggregates within neurons, astrocytes, and oligodendrocytes, leading to progressive neurodegeneration. Normally, tau protein is highly expressed in axons as it is required for microtubule stability and assembly.[26] However, hyperphosphorylation changes conformation and reduces its binding affinity for microtubules, impairing the latter's function. Dissociated tau has a higher propensity for multimerization, aggregation, and contributing to neurodegeneration.[4] The neurodegenerative process appears to spread in a prion-like manner across synapses, potentially explaining disease progression.[27]
The resultant tau inclusions disrupt neuronal function, contributing to synaptic dysfunction, axonal transport failure, and cell death. In support of this argument, results from a mouse-model-Alzheimer disease study showed that tau triggers N-methyl-D-aspartate (NMDA) receptor-mediated excitotoxic signaling via targeting the postsynaptic Src kinase Fyn.[28] In addition, tau also disrupts synaptic function by accumulating in dendritic spines and reducing the count of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and NMDA receptors.[29]
Early pathological involvement is seen in the striatum and anterior frontal regions, with posterior spread over time. In advanced cases, widespread tau pathology affects the frontal, parietal, and temporal cortices, striatum, and corpus callosum.[30] Clinically, CBS is linked to perirolandic pathology. Still, other neurodegenerative diseases such as Alzheimer disease, PSP, Pick disease, Creutzfeldt-Jakob disease, and transactive response deoxyribonucleic acid binding protein-43 (TDP-43) proteinopathies can also underlie CBS.[31][32]
Beyond tauopathy, the neurodegenerative cascade in CBD involves mitochondrial dysfunction, impaired axonal transport, inflammation, and synaptic degeneration. Neuroinflammation is a key driver, with activated microglia and astrocytes releasing proinflammatory cytokines, including tumor necrosis factor-alpha, interleukin (IL)-1 beta, IL-6, and complement proteins. Microglia also facilitate tau propagation through endocytosis and exocytosis cycles.[1] Elevated levels of chitinase-3-like protein 1, a marker of astroglial neuroinflammation, have been observed in CBD and other tauopathies.[33]
White matter degeneration is another significant pathological feature, likely stemming from tau-mediated oligodendrocyte dysfunction, contributing to subcortical rarefaction and U-fiber loss. The asymmetric distribution of cortical and basal ganglia involvement correlates with the asymmetric clinical presentation of motor and cognitive deficits. Results from genetic studies have linked CBD to the H1 haplotype of the MAPT gene, which increases the risk of tau dysfunction and aggregation.
Additional genome-wide association study results have implicated myelin-associated oligodendrocyte basic protein and son of sevenless homolog 1, highlighting a potential role for white matter pathology and non-tau-mediated mechanisms. There is also emerging evidence of genetic overlap between CBD and immune-mediated diseases, suggesting a possible role for immune system dysregulation. Despite these advances, the precise triggers of tauopathy and the full extent of pathogenic pathways in CBD remain incompletely understood, underscoring the need for further research to facilitate targeted therapeutic development.
Histopathology
The histopathology of CBD is defined by distinct neuronal, glial, and axonal changes underpinned by hyperphosphorylated 4R tau protein deposition (see Image. Histopathologic Characteristics of Corticobasal Degeneration). These histologic features are essential for distinguishing CBD from other tauopathies, guided in part by the "Office of Rare Diseases Neuropathologic Criteria for CBD" (2002), which provides a standardized framework for diagnosis. At minimum, CBD diagnosis requires tau-positive neuronal and glial lesions in the cortex and striatum, particularly astrocytic plaques, thread-like lesions in white and gray matter, and focal neuronal loss in the cortex and substantia nigra.
Core Histopathological Features
- Astrocytic plaques
- Astrocytic plaques are the hallmark lesion of CBD. These are formed by the deposition of hyperphosphorylated 4R tau within astrocytic processes, typically observed in the cortical gray matter and striatum. These plaques coexist with tau-laden dendritic neurites and oligodendroglial cytoplasmic tau tangles.[34][35] Tau deposition within glial cells contributes to widespread neuroinflammation and axonal dysfunction.
- Ballooned neurons
- Ballooned neurons, or achromatic neurons, are a key histologic feature of CBD. These swollen neurons, predominantly found in the third, fifth, and sixth cortical layers, contain hyperphosphorylated tau aggregates.[36] Their distribution correlates with cortical atrophy and localized functional impairments, particularly in motor and sensory regions.
- White matter rarefaction
- Subcortical white matter, including U-fibers, exhibits significant rarefaction in CBD.[37] This reflects widespread axonal degeneration and loss of connectivity between cortical and subcortical structures, contributing to the clinical manifestations of CBS.
- Thread-like lesions
- Tau-positive thread-like lesions in white and gray matter are a defining feature of CBD. These thread-like structures are associated with disrupted neuronal processes and impaired synaptic connectivity.
- Neuronal loss and spongiosis
- Neuronal loss is focal, most prominent in the substantia nigra and the affected cortical regions. Microvacuolization or laminar spongiosis may be present, particularly in the superficial cortical layers, although this finding can extend across all layers.
Subtypes of CBD Pathology
The distribution and severity of tau lesions allow for classification into 3 pathological subtypes:
- Typical CBD
- Predominant involvement of the posterior frontoparietal or perisylvian cortex
- Basal ganglia-predominant CBD
- Severe degeneration in the pallidum and subthalamic nucleus with milder cortical involvement
- PSP-like CBD
- Severe brainstem and dentate nucleus degeneration, with variable cortical involvement
Tau Pathology in Clinical Variants
CBD manifests primarily as 2 clinical syndromes, CBS (CBD-CBS) and PSP-like syndrome (CBD-PSPS), each with distinct patterns of tau deposition:
- CBD-CBS
- There is a greater tau burden in the primary motor and somatosensory cortex, subthalamic nucleus, putamen, and globus pallidus externa. Neuronal loss in the medial substantia nigra is less severe.
- CBD-PSPS
- The tau burden is more extensive in the hippocampus, cerebellum, brainstem white matter tracts, and superior frontal cortex. Patients exhibit greater neuronal loss in the substantia nigra and more prominent tau pathology in limbic regions, such as the anterior thalamus and hippocampus.[38]
Prion-Like Tau Propagation
A prion-like spread of tau across synapses has been implicated in the pathogenesis of CBD. Misfolded tau aggregates propagate from cell to cell, seeding further tau pathology. This process exacerbates neurodegeneration and explains the progressive and asymmetric distribution of lesions.
Inflammatory and Synaptic Mechanisms
Several mechanisms mediate tau-induced neurodegeneration:
- Microtubule dysfunction
- Hyperphosphorylated tau dissociates from microtubules, impairing their stability and assembly. The dissociated tau aggregates form neurotoxic filaments.
- Synaptic dysfunction
- Tau disrupts synaptic function by targeting NMDA and AMPA receptors, leading to excitotoxicity and dendritic spine loss.
- Neuroinflammation
- Tau activates microglia and astrocytes, triggering proinflammatory cytokines like tumor necrosis factor-α, IL-1β, and IL-6. Microglia may also mediate tau spread through cycles of endocytosis and exocytosis.
Additional Pathological Features in CBD-PSPS
Patients with CBD-PSPS often show additional pathologies, such as TDP-43 inclusions, particularly in the midbrain tegmentum.[39] These inclusions contribute to the more severe cognitive and behavioral impairments seen in this subtype compared to CBD-CBS.
Pathological Insights and Diagnosis
Immunohistochemical staining for hyperphosphorylated tau is crucial for diagnosing CBD. Astrocytic plaques, ballooned neurons, and tau-positive thread-like lesions distinguish CBD from other tauopathies. However, additional clinical or genetic data may be needed to differentiate CBD from frontotemporal dementia and Parkinsonism linked to chromosome 17.
History and Physical
History
CBD is a progressive neurodegenerative disorder with insidious onset, typically beginning in the sixth or seventh decade of life. The initial presentation varies among patients but often includes asymmetric motor dysfunction, cognitive impairment, or language deficits. The disease follows a relentlessly progressive course over several years, leading to severe disability.
In 2013, Armstrong et al and an international consortium of behavioral neurologists, neuropsychologists, and movement disorder specialists published updated clinical criteria for the diagnosis of CBD. These criteria were developed in response to the growing understanding of CBD and its clinicopathologic correlations based on 267 nonoverlapping pathologically confirmed CBD cases. From this effort, 4 distinct clinical phenotypes of CBD were identified:
- Probable and possible CBS
- Frontal-behavioral and spatial syndrome (FBS)
- A nonfluent agrammatic primary progressive aphasia syndrome (nfa-PPA)
- PSPS
Clinical Criteria for CBD
The clinical phenotypes were integrated to establish 2 overarching diagnostic categories:
- Probable CBD
- Requires an age of onset of more than 50 years
- Excludes the presence of a family history or a known genetic mutation affecting tau protein
- Diagnosis can only be made with:
- Probable CBS, or
- FBS or nfa-PPA, provided at least 1 CBS feature (i-vi) is present [16]
- Possible CBD
- Does not require a specific age of onset
- Allows for a positive family history or a known tau-protein mutation
- Diagnosis can be made with:
- Possible CBS, or
- FBS or nfa-PPA, or
- PSPS, provided at least 1 CBS feature (ii-vi) is present
Clinical Criteria for CBS
CBS is also divided into 2 overarching diagnostic categories:
- Probable CBS
- Characterized by an asymmetric presentation of at least 2 of the following:
- Limb rigidity or akinesia
- Limb dystonia
- Limb myoclonus
- Plus, at least 2 of:
- Orobuccal or limb apraxia
- Cortical sensory deficit
- Alien limb phenomenon (must be more than simple levitation)
- Plus, at least 2 of:
- Characterized by an asymmetric presentation of at least 2 of the following:
- Possible CBS
- May present symmetrically and requires 1 of the following:
- Limb rigidity or akinesia
- Limb dystonia
- Limb myoclonus
- Plus, at least 1 of:
- Orobuccal or limb apraxia
- Cortical sensory deficit
- Alien limb phenomenon (more than simple levitation)
- Plus, at least 1 of:
- May present symmetrically and requires 1 of the following:
Limitations of the Clinical Criteria
Study results have indicated that these diagnostic criteria lack specificity. No single clinical feature can reliably distinguish CBD from other neurodegenerative pathologies, such as Alzheimer disease (AD) or PSP, in patients presenting with CBS.[16][40] This diagnostic overlap underscores the complexity of CBD and its clinical mimics.
Pathologic Heterogeneity of CBS
To emphasize the variability of underlying pathology in CBS, a review of the Mayo Clinic Brain Bank until November 2021 of 345 brains from patients with an antemortem diagnosis of CBS recognized the underlying pathologic diagnoses to be of CBD (32%), PSP (31%), AD (20%), and others (17%), including diffuse Lewy body disease, frontotemporal lobar degeneration with TAR deoxyribonucleic acid-binding protein 43 (FTLD TDP), motor neuron disease, Pick disease, frontotemporal lobar degeneration with fused-in-sarcoma pathology (FTLD FUS), frontotemporal lobar degeneration with tau pathology (FTLD tau), Creutzfeldt-Jakobs disease, and cerebrovascular disease. CBS, regardless of the underlying pathology, has frontoparietal, as well as motor and premotor involvement.[37]
Physical
The physical examination of a patient with CBD reveals asymmetrical motor and cognitive impairments, as well as cortical sensory deficits and speech disturbances, which further distinguish CBD from other neurodegenerative disorders. The presentation can be highly variable, reflecting the heterogeneity of its underlying neuropathology. While asymmetrical rigidity, dystonia, myoclonus, and apraxia are hallmark features, some patients may present with predominant language dysfunction, cognitive impairment, or corticospinal signs. This variability often leads to diagnostic challenges and overlaps with other neurodegenerative disorders.
Clinical Features of CBD
The most common clinical presentation of CBD is corticobasal syndrome (CBS), a clinical syndrome characterized by cortical and extrapyramidal signs. The most common cortical signs are apraxia, cortical sensory deficits, and alien limb phenomenon, and the most common extrapyramidal signs are parkinsonism, dystonia, and myoclonus.[23] Common physical exam findings include:
- Parkinsonian features and responsiveness to levodopa
- Characteristically, parkinsonism in CBS is unilateral or asymmetric; however, symmetric CBD has been reported. This symmetric form of CBD is rare and typically associated with younger age at onset (median 61 vs 66, P < 0.05), less asymmetric cortical imaging findings, with a relative absence of other features of CBS such as myoclonus, dystonia, alien limb phenomenon, and limb apraxia.[41] Asymmetric limb rigidity and bradykinesia are likely the most common parkinsonian manifestations in pathologically proven CBD.[16][22] Limb rigidity may be lead-pipe in nature with or without cogwheeling and may have a coexistent component of dystonia and gegenhalten/paratonic rigidity in cases with predominant frontal lobe dysfunction.[16] As the disease progresses, limb rigidity involves all limbs.[22] Axial rigidity is less common in CBD as compared to other atypical parkinsonian disorders such as PSP.[22] This sign has been reported in about 27% of the cases at presentation and 69% at some point of disease evolution.[16]
- Levodopa response can be moderate in a minority of patients; however, it is only transient. Levodopa-induced dyskinesias, akin to Parkinson disease (PD), have also been described.[42] Results from a study of 147 patients with CBD found that about one-fourth of the patients demonstrated at least moderate improvement in their parkinsonian symptoms attributable to levodopa; however, only 7 patients were autopsy-proven, and the retrospective nature of the study also limited the information on the magnitude or duration of the response.[24]
- Tremor is less frequent in CBD than in PD and is phenotypically distinct from the PD's tremor.[24] Tremor has been described in about 39% of patients with CBD at some point in their disease course. This type of tremor usually combines resting, positional, and action tremor, irregular and sometimes resembling low-amplitude action myoclonus.[16]
- Gait difficulties, postural instability, and falls
- Gait problems can present in about 73% of patients with CBD throughout their disease course.[16] Gait can present similarly to that in PD with features of bradykinesia, shuffling, and decreased arm swing; however, a wider-based gait with freezing has also been reported.[2] Freezing of gait tends to happen with increased disease duration, with an incidence of about 1 out of 13 patients within 3 years from disease onset and a 3-fold increase by 6 years.[43] Nonetheless, CBD should not typically present as a primary progressive freezing gait disorder.[44] Results from a study suggested that the frequency of falls and gait changes in patients with CBD-CBS are 29% and 36%, respectively, at first evaluation, but this seems to be an exception rather than a rule for the disease.[16] Apraxia can also manifest with gait abnormalities and falls.[2]
- Dystonia
- According to large reviews of pathologically confirmed cases, dystonia appears to be present in about 40% of patients with CBD.[16][45] However, some study results have shown dystonia to be as frequent as in 90% to 100% of cases, perhaps due to referral bias and the inclusion of nonpathologically confirmed cases.[46][47]
- Dystonia typically occurs within 2 years from disease onset and commonly affects the upper limb. Dystonia may evolve into hemidystonia or affect the other side, but it rarely starts in the leg.[45] Classic dystonic presentations are of adduction and flexion at the arm, forearm, wrist, and metacarpophalangeal joints with extension at interphalangeal joints, as opposed to flexion and internal rotation at the hip with flexion at the knee and inversion at the foot.[47] Interestingly, the onset of dystonia was later in the disease course in the cases with a "dementia" (frontotemporal dementia or Alzheimer disease) phenotype. The study also hinted at a possible association between dystonia and myoclonus as they almost cooccurred in their cases.[45]
- Myoclonus
- Myoclonus has been estimated to be prevalent in about 27% of cases during the entire disease course.[16] Myoclonus has been described to be primarily focal, affecting the upper extremities and less so the face, more prominent in response to sensory stimulation and on voluntary action, ie, stimulus-induced, and at times to be superimposed with limb dystonia.[48][49][50][51] The known cortical pathology in CBD and the focal, predominantly distal, hypersynchronous jerks, with evidence of cortical hyperexcitability via magnetic brain stimulation in a study, suggest that the myoclonus in these patients is likely cortical in origin. Electrophysiologically, the myoclonus of CBD differs from cortical reflex myoclonus due to shorter latency of reflex jerks (40 ms vs 50 ms in hand muscles), absence of enlargement of cortical sensory evoked potentials, and lack of preceding cortical discharge in the back-averaged electroencephalogram before each jerk. These differences are hypothesized to arise from the enhanced direct sensory input to the motor cortex in CBD, as opposed to abnormal direct relays through the sensory cortex to the motor cortex or via cerebellar-thalamocortical projections in cortical reflex myoclonus. The specific electrophysiologic finding in favor of the above hypothesis is that the latency of reflex myoclonus in CBD is only 1 to 2 ms more than the sum of the afferent and efferent times to and from the cortex.[50]
- Bulbar symptoms
- Results from a clinical study of 36 CBD patients (only 6 underwent neuropathological studies) revealed that dysarthria was present in 11% of patients early in the disease course, and the number rose to 70% at the 5-year follow-up.[52]
- The median latencies of dysarthria and dysphagia were estimated to be around 40 months and 64 months, respectively, by a postmortem study of 13 patients with CBD in comparison to patients with PD and other APDs such as dementia with Lewy bodies, PSP, and multiple system atrophy. Similar to other parkinsonian disorders, survival decreases with the onset of dysphagia, with a median latency of 15 to 24 months. Therefore, early identification and treatment of dysphagia is fundamental to prevent or delay complications such as aspiration pneumonia and, in turn, potentially improve quality of life and increase survival time.[53]
- Apraxia
- Apraxia is the clinical hallmark of CBS and presents at disease onset in about 45% of patients and later in the disease course in around 57% of patients.[16] Assessing praxis in the less affected limb may be helpful to avoid inaccurate assessment secondary to concomitant motor symptoms (like rigidity and dystonia) or involuntary movements (like myoclonus). Typically, it starts in the upper extremities and is of the ideomotor type (not knowing "how to do it"). However, the advanced stages of CBD can manifest ideational apraxia (not knowing "what to do"), which is less commonly seen otherwise in the disease.[54][55] The same applies to orobuccal apraxia, which tends to appear later in the disease course, although it can rarely be the initial manifestation of concurrent loss of speech output.[56] Ideomotor apraxia is associated with a decreased gray matter volume within the left supplementary motor area, premotor cortex, and caudate nucleus of patients with CBS.[57] In patients with CBS with more severe apraxia (usually a combination of ideomotor and ideational apraxia), there often is a global cognitive impairment and additional parietal or diffuse cortical damage.[54] The degree of apraxia appears independent of the side of motor impairment. Transitive and intransitive praxis seem to be equally impaired; however, praxis to imitating gestures seems to be worse impacted than to commands.[57]
- Aphasia
- Language impairments are reported in about 53% of patients with CBD. They have been described with a broad spectrum ranging from mild abnormalities to severe progressive nonfluent aphasia, with some even progressing to complete mutism. Aphasia has primarily been found to be of anomic, Broca, or transcortical types but not receptive or Wernicke. These types are associated with left frontoparietal cortical damage and abnormalities within the subcortical white matter and corpus callosum.[16][25][58][59] Additionally, apraxia of speech, defined as problems with translating conscious speech plans into motor plans presenting with characteristic articulation and prosody errors, has been described in CBD as coexistent or independent from aphasia.[38]
- Results from a retrospective study on 38 individuals with CBS established an association between aphasia and right-sided motor symptoms, further confirming that it stems from left hemispheric dysfunction. Such an association did not exist in patients with left-predominant motor presentation. Additionally, dysarthria did show such a preferential correlation.[60]
- Alien limb phenomenon
- At the time of its initial description, the alien limb phenomenon was primarily thought of as a subjective difficulty in recognizing one's limb as one's own, especially in the absence of visual input.[61] Today, the alien limb implies one that carries "a will of its own" or "is foreign" together with an observable motor activity. Thus, it can be defined as an involuntary movement in the context of feeling estranged from the limb. The limb may perform meaningful acts without being guided by the patient's intention and may reach and grab objects with the inability to release grasped objects without intervention from the unaffected extremity; patients often express surprise and frustration at this errant limb and refer to it in the third person.[62][63][64]
- This phenomenon is present in about 30% of cases at any point during the disease course. The alien limb is usually localized to the corpus callosum alone, the corpus callosum and the dominant medial frontal cortex, or the posterior cortical/subcortical region. The consensus, however, is that the alien limb movements in CBD tend to be different than in patients with frontal lobe or corpus callosum lesions. Perseverative movements are less common, especially early in the disease's course. The alien limb is likely to drift, levitate, or assume odd postures similar to the patterns seen in parietal lesions. These movement patterns are presumed to stem from interactions between frontal lobe-mediated "avoidance" behaviors and parietal lobe-mediated "approach" behaviors. While frontal lobe damage may release parietal "approach" behaviors, parietal lobe injuries may release frontal "avoidance" behaviors, resulting in abnormal, everted, and overextended postures.[64][65] In line with this frontal-parietal lobe interaction, a bilateral asymmetric alien limb syndrome has also been reported in CBD, where 1 hand demonstrated tactile avoidance with levitation from presumed parietal dysfunction, and the other hand showed a continuous tactile pursuit of the examiner's hand from presumed frontal dysfunction.[66]
- Other
- Apart from the well-established clinical phenotypes mentioned above, there are other potential manifestations of the disease:
- AD-like amnestic phenotype [59][67]
- Conduction-type aphasia with prominent impairment in repetition [68]
- Posterior cortical atrophy with Balint syndrome triad of optic ataxia, oculomotor apraxia, and simultagnosia [69]
- Sleep disturbances including rapid eye movement, sleep behavior disorder, and insomnia [70]
- Neuropsychiatric complaints like apathy, depression, agitation, or impulsivity [71][72]
- Autonomic dysfunction with urinary incontinence/constipation [71][73]
- Progressive oculovisual dysfunction [74]
- Frontal-type gait disorder [75]
- Apart from the well-established clinical phenotypes mentioned above, there are other potential manifestations of the disease:
Evaluation
There are no definitive laboratory tests for CBD, but a comprehensive workup is essential to rule out alternative diagnoses and identify supportive biomarkers.
Laboratory Tests
Blood Tests
Routine blood tests are typically unremarkable in CBD but are performed to exclude other causes of parkinsonism and cognitive impairment, including:
- Thyroid function tests
- To rule out hypothyroidism-related cognitive and motor dysfunction
- Vitamin B12, folate, and homocysteine levels
- To assess for vitamin deficiencies contributing to neurodegenerative symptoms
- Syphilis and human immunodeficiency virus serology
- To rule out infectious causes of dementia and movement disorders
- Autoimmune and paraneoplastic panels
- In cases with atypical presentations to exclude autoimmune encephalitis and paraneoplastic syndromes
- Genetic testing
- If there is a strong family history, testing for MAPT mutations or other tauopathies may be considered, though CBD is primarily sporadic.
Cerebrospinal Fluid Biomarkers
Cerebrospinal fluid (CSF) biomarkers have emerged as promising tools for diagnosing CBD, particularly for distinguishing it from other neurodegenerative disorders. Given the central role of tau pathology in CBD, recent research has focused on identifying isoform-specific tau species that reflect disease-specific neurodegeneration. Measuring soluble tau fragments in CSF can provide insights into the underlying pathological process, aiding in the early and accurate identification of CBD.
Results from a recent study on 4R isoform-specific tau species from microtubule-binding region (MTBR), ie, MTBR-tau275 and MTBR-tau282, showed that when normalized to total-tau, they increase in brain insoluble tau and decrease in the CSF soluble tau in primary tauopathies, especially CBD and FTLD-MAPT P301L. Interestingly, CSF MTBR-tau275/t-tau and MTBR-tau282/t-tau did not decrease in PSP and argyrophilc grain disease (AGD), the other 4R-tauopathies. This discrepancy originated from variability in the degree of neocortical pathology in PSP and AGD cases compared to CBD and FTLD-MAPT cases. This study, thus, proposes the potential role of CSF MTBR-tau275 and MTBR-tau282 as the first affirmative fluid disease biomarkers for primary tauopathies, especially CBD and FTLD-MAPT.[76]
CSF biomarkers for Alzheimer disease (AD), such as amyloid beta 42, total tau (t-tau), and phosphorylated tau, are crucial in ruling out AD in patients with overlapping clinical features. In a patient with predominantly cognitive symptoms, the possibility of underlying AD pathology should be considered first before considering other differentials like CBD, and this can be ascertained with the CSF levels of amyloid beta 42, t-tau, and p-tau with a sensitivity of 95% and specificity of 83%.[77] Additionally, neurofilament light chain (NfL) may be elevated in CBD, reflecting neurodegeneration.
Neuroimaging Studies
Radiographic imaging is critical for supporting the diagnosis of CBD and distinguishing it from other neurodegenerative conditions.
Magnetic Resonance Imaging
Magnetic resonance imaging is a valuable tool in the evaluation of CBD, aiding in the exclusion of other neurodegenerative disorders and identifying characteristic atrophy patterns. Findings often include asymmetric cortical atrophy, particularly in the parietal and frontal lobes, and basal ganglia involvement (see Image. Corticobasal Degeneration on Magnetic Resonance Imaging). However, it is important to note that the imaging finding of asymmetric frontoparietal atrophy is more reflective of a clinical syndrome of CBS than an underlying CBD pathology. Advanced imaging techniques like diffusion tensor imaging and volumetric analysis may enhance diagnostic accuracy by detecting early structural and functional changes. However, MRI findings are often nonspecific, necessitating clinical correlation for definitive diagnosis.
A voxel-based morphometry study to analyze atrophy patterns found that patients with a clinical syndrome consistent with CBS exhibited perirolandic atrophy involving both the anterior and posterior central gyrus, regardless of the underlying pathology. In contrast, patients with pathologically confirmed CBD demonstrated atrophy in the perirolandic region and the dorsal prefrontal cortices, striatum, and brainstem.[78] Results from another study examining gray matter thinning in CBS patients revealed consistent involvement of the premotor cortex and insula in the dominant hemisphere and the supplementary motor area bilaterally. However, the distribution of atrophy varied based on the underlying pathology. Specifically, in CBD-CBS cases, gray matter loss extended from the premotor cortex into the inferior and superior posterior frontal lobes and supplementary motor area, a pattern closely resembling that observed in PSP-CBS, except for relative sparing of the inferior frontal lobe.[79]
Additionally, a correlation study using MRI volumetric and diffusion tensor imaging (DTI) in 47 patients (28 PSP and 19 CBD) indicated that 'brain volume' and 'white matter integrity' might serve as useful surrogate markers for tau pathology in 'subcortical/brainstem' and 'cortical' regions, respectively.[80] Results from another diffusion tensor imaging study involving 11 patients with probable CBD reported an increased apparent diffusion coefficient average in the motor thalamus and the precentral and postcentral gyri ipsilateral to the affected frontoparietal cortex, as well as in the bilateral supplementary motor area. Fractional anisotropy values were predominantly reduced in the precentral and supplementary motor areas, followed by the postcentral gyrus and cingulum.[81] However, a significant limitation of this study was the absence of pathological confirmation of CBD in these patients.
Positron Emission Tomography
Positron emission tomography (PET) imaging is valuable in evaluating CBD, providing metabolic and molecular insights that complement clinical and structural findings. PET imaging utilizes radioligands to bind to in vivo targets to assist in quantitative visualization of functional processes. The most used radiotracer is [18F] fluorodeoxyglucose (FDG), which helps assess regional brain glucose metabolism. Over the past decade, various pathology-specific radiotracers, such as amyloid and tau tracers, have been developed to image tauopathies, especially AD, with a growing literature in 4R-tauopathies.
Fluorodeoxyglucose-Positron Emission Tomography
FDG-PET plays a crucial role in identifying regional differences in brain metabolism, which can aid in differentiating neurodegenerative disorders, particularly in the early stages when clinical distinctions between diseases remain challenging.[82] FDG-PET detects asymmetric cortical hypometabolism, particularly in the parietal and frontal lobes, aiding in differentiation from other neurodegenerative disorders. Results from a study identified and validated a distinct metabolic covariance pattern in 10 patients diagnosed with probable CBD. This pattern was characterized by asymmetric bilateral hypometabolism in the cerebrum, specifically involving the frontal and parietal cortices, thalamus, and caudate nucleus, with a relative increase in metabolism observed in the occipital regions. However, this study had notable limitations: (1) all participants exhibited the CBS phenotype, which includes asymmetric limb involvement and apraxia, meaning the identified metabolic pattern may not be representative of other CBD presentations; and (2) only 3 of the patients had a pathologically confirmed CBD diagnosis, raising the possibility that some may have had a different underlying pathology.[83]
Addressing these limitations, another study examined 29 CBS patients who underwent FDG-PET imaging and postmortem neuropathological evaluation. Of these, 14 were confirmed to have primary CBD pathology (CBD-CBS). Hypometabolism patterns varied across different underlying pathologies, but CBD-CBS patients demonstrated a more pronounced bilateral basal ganglia involvement. Notably, the primary motor cortex was the only common hypometabolic region among all groups. Based on these results, the study suggested that FDG-PET could be valuable for differentiating pathological diagnoses in patients with CBS.[84] Further research is needed to determine its applicability in CBD cases that do not present with the CBS phenotype.
Tau PET Imaging
Tau PET imaging has emerged as a promising technique for assessing tau pathology in neurodegenerative diseases. Among the first-generation tracers, [18F]AV-1451 (flortaucipir) demonstrated increased uptake in the bilateral premotor and motor cortices and globus pallidus in patients with CBD patients compared to controls. Additionally, there was a trend toward greater globus pallidus uptake in patients with CBD compared to PSP, suggesting potential utility in differentiating tauopathies. However, [18F]AV-1451 has significant limitations, including poor specificity for 4R tau, the dominant tau isoform in CBD, as it preferentially binds to the mixed 3R/4R paired helical tau filaments found in AD.[85][86] Furthermore, off-target binding remains a challenge, particularly due to the tracer's high affinity for monoamine oxidase-B (MAO-B), complicating interpretation.[87] Results from another study investigating antemortem tau PET imaging in relation to postmortem autopsy findings demonstrated that while [18F]AV-1451 reliably identified advanced Braak tau pathology in AD, it failed to detect early-stage neurofibrillary tangle pathology and was unable to differentiate non-AD tauopathies, including CBD.[88]
To overcome these limitations, second-generation tau tracers have been developed. [18F]PM-PBB3 (also known as [18F]-APN-1607) has shown greater efficacy in binding to and tracking the accumulation of 4R tau over time, particularly in carriers of MAPT mutations.[89] Results from a case study involving a patient with auditory agnosia and dysprosody, [18F]PM-PBB3 uptake was observed in the occipital cortex, asymmetrical neocortex, subcortical regions, basal ganglia, thalamus, and midbrain. Based on this distribution, the authors suspected underlying CBD pathology; however, the absence of pathological confirmation limited definitive conclusions.[90]
Another promising tracer, [18F]PI-2620, has shown asymmetric pallidal uptake in patients with CBD, distinguishing it from patterns observed in PD and PSP.[91] A study examining patients with CBS provided results demonstrating that there was preferential uptake in the dorsolateral prefrontal cortex and basal ganglia contralateral to the clinically affected side, irrespective of beta-amyloid status. This suggests that [18F]PI-2620 may be valuable in assessing underlying pathology and tracking disease progression in CBS.[92]
Despite the advances in tau PET imaging, significant challenges remain, particularly concerning off-target binding and inconsistencies between antemortem imaging and postmortem pathological findings. Although second-generation tracers like [18F]PM-PBB3 and [18F]PI-2620 have shown improvements in reducing off-target binding in the basal ganglia, cerebral white matter, and monoamine oxidases-A/B, they have introduced new binding artifacts in regions such as the skull and meninges. Furthermore, the specificity of these tracers for 4R tau requires further validation through larger-scale studies.[85]
Other Diagnostic Tests
Other diagnostic tests can aid in differentiating CBD from other neurodegenerative conditions. Dopamine transporter imaging (eg, DaTscan/SPECT) may reveal asymmetric striatal dopamine transporter loss; however, this finding is nonspecific and can also be observed in other parkinsonian syndromes. Electromyography and nerve conduction studies are often performed to exclude mimicking conditions such as motor neuron disease. Additionally, neuropsychological testing assesses cognitive function in patients with CBD, identifying deficits that vary based on disease phenotype. Common findings include executive dysfunction, apraxia, nonfluent aphasia, and visuospatial impairments.
Treatment / Management
Symptomatic Management
The treatment framework below is largely derived from the CurePSP Centers of Care consensus on best practices in PSP and CBS clinical management.[71] However, given the large clinical heterogeneity in the disease presentation and individual disease manifestations, a tailored care plan must be synthesized for individuals with CBD with the help of an interprofessional team including neurologists, psychotherapists, speech therapists, psychiatrists, ophthalmologists, sleep specialists, occupational therapists, physical therapists, and palliative care physicians.(B3)
A levodopa trial should be conducted for parkinsonism. An adequate trial would entail titrating it over a month to 900 to 1200 mg daily in 3 divided doses and maintaining the maximum tolerated dose for at least a month. If there is no response or the emergence of adverse events like dyskinesias, the medication must be tapered slowly over at least 2 weeks. Abrupt discontinuation can cause parkinsonism-hyperpyrexia syndrome. Other dopaminergic agents like monoamine oxidases-B inhibitors or catecholamine O-methyltransferase inhibitors are even less likely to provide benefits than levodopa.
Constipation and urinary disturbances may become prominent as a part of possible autonomic dysfunction in CBD. Constipation may respond to increased physical activity, adequate hydration, fiber supplementation, and pharmacologic therapy with stool softeners or osmotic laxatives. Stimulant laxatives should only be used short-term. Medications like guanylate cyclase c agonists (linaclotide) and selective 5-hydroxytryptamine receptor 4 agonists (prucalopride) may need to be prescribed in refractory cases of constipation.
Urinary dysfunction may be targeted with the use of alpha-receptor antagonists (tamsulosin, terazosin, doxazosin, alfuzosin, silodosin), 5-alpha reductase inhibitors (finasteride, dutasteride), beta-3 adrenoceptor agonists (mirabegron, vibegron), and selective M3 antimuscarinic anticholinergics (darifenacin, solifenacin); however, nonselective antimuscarinic agents (oxybutynin, tolterodine, fesoterodine) should be avoided given central anticholinergic side effects. Botulinum toxin injections may need to be considered for refractory overactive bladder. Polypharmacy can be avoided using nonpharmacologic measures like condom catheters for men, clean intermittent urinary catheterization, and transtibial nerve stimulation.
Melatonin can manage sleep disturbances like insomnia and rapid eye movement sleep behavior disorder; however, low-dose clonazepam may be necessary if melatonin fails. Given cognitive side effects, other benzodiazepines, benzodiazepine receptor agonists, and tricyclic antidepressants must be avoided. Stimulants, if used for excessive daytime somnolence, must be restricted to low doses.
As bulbar symptoms progress, sialorrhea and dysphagia may become more troublesome. Botulinum toxin is the first-line therapy for sialorrhea. Sublingual atropine drops are not recommended due to central anticholinergic adverse events, and oral glycopyrrolate, if used, should be monitored for the same reason. In cases of dysphagia, an early referral to speech-language pathology should be placed. Conservative measures with small bites and sips, multiple swallows, and thickened liquid consistencies can be helpful; however, percutaneous gastrostomy tube placement may become necessary in severe cases, necessitating timely goals-of-care discussions by the treating physician.
Apraxia of speech and nonfluent aphasia are best managed by intense, repetitive exercises and using gestures, nonspeech modalities, and pacing methods with the help of a trained speech pathologist. Transcranial magnetic stimulation to the cerebellar region and transcranial direct current stimulation to the dorsolateral prefrontal region are potential experimental treatments for speech apraxia and aphasia, respectively. Limb apraxia warrants occupational therapy with interventions like gestural training, bimanual or bipedal tasks, mirror therapy, repetitive facilitation exercises, or video game-based rehabilitation.
If focal, botulinum toxin is the best treatment for dystonia. Other pharmacologic therapies include baclofen and clonazepam. Baclofen can be started at 5 mg daily and titrated to a maximum dose of 10 mg 3 times a day, while clonazepam is usually started at 0.25 mg daily and titrated to no more than 3 mg per day. Occupational therapy with an evaluation for orthoses may be beneficial for dystonia.
Pharmacologic management of cognitive impairment with cholinesterase inhibitors should be reserved for amnestic deficits. Caregiver and family education, establishment and maintenance of a daily routine, occupational therapy, external memory aids (eg, calendars, daily planners), technology (eg, mobile devices), and lifestyle modifications may be beneficial nonpharmacologic measures. Behavioral symptoms of apathy can be managed with a trial of methylphenidate or modafinil; depression and other mood disorders may respond to selective serotonin reuptake inhibitors (SSRIs). Impulsivity, if present, may need reduction of levodopa, mood stabilizers, a trial of SSRIs, or mediations used for attention deficit hyperactivity disorder like atomoxetine. Progressive eyelid and visual dysfunction may lead to blepharospasms, decreased blinks, reduced tear production, diplopia, saccadic slowing, and hypometria. Botulinum toxin for blepharospasm, artificial tears for dry eyes, binocular prisms for gaze limitation, environmental modifications, and referral to an ophthalmologist or a neurooptometrist may all prove helpful in the symptomatic management of ocular and visual dysfunction.
Targeted Tau Therapy
With the recent landmark United States Food and Drug Administration approval of drugs targeting amyloid pathology, there has been an increased interest in developing monoclonal antibodies targeting intracranial pathologies such as amyloid beta and tau.[93][94] However, a similar breakthrough has not yet been achieved with the advent of tau-directed therapies.[95] Therefore, it has become more pertinent than ever to develop disease-specific biomarkers to help predict the pathology in the antemortem diagnosis of neurodegenerative diseases with increased sensitivity and specificity and monitor the quantitative effect of the treatment over time.(A1)
Clinical trials in developing a disease-modifying therapy for 4R-tauopathies either focus on reducing the toxic gain of function caused by pathologic tau aggregates or restoring a loss of normal tau function.[96] Toxic gain of function may be mitigated by reducing tau gene expression, preventing posttranslational modifications that promote tau aggregation, or clearing tau aggregates mostly via an immunologic mechanism. Loss-of-function restoration strategies focus on microtubule stabilization.[97]
An immunoglobulin G4 monoclonal antibody, gosuranemab, against the N-terminal region of tau reduced both full-length tau and extracellular tau fragments in cell culture and animal models; however, its results showed a failure in demonstrating efficacy in the phase 2 PASSPORT study in PSP-Richardson syndrome (PSP-RS) patients.[98] As a result, a parallel basket study, TauBasket, examined other 4R-tauopathies like CBD, nonfluent variant primary progressive aphasia, traumatic encephalopathy, and frontotemporal lobar degeneration-MAPT was terminated due to futility analysis results from PASSPORT.[97] A major controversy in the use of monoclonal antibodies surrounds the appropriate tau protein epitope target, and it continues to be a matter of ongoing investigation. A transgenic mice model showed that antibodies targeting the tau's middomain region suppressed tau aggregation, whereas N-terminal antibodies did not.[99] A tau middomain monoclonal antibody, UCB0107, completed phase 1 studies in health controls, and an open-label extension study is ongoing.[97]
A microtubule stabilizer, TPI 287, was evaluated in AD and 4R-tauopathies, PSP, and CBS; however, it showed a dose-related worsening in cognitive outcomes in the 4R-tauopathy arm, preventing further development.[100] Most other trials have primarily been conducted in individuals with PSP-RS. Neuroprotective drugs (riluzole, rasagaline), tau phosphorylation inhibitors (glycogen synthase kinase-3 beta inhibitors like lithium, valproate, tideglusib), and tau acetylation inhibitors (salsalate) in PSP-RS have largely produced discouraging results. (A1)
Other prospective potential agents in various stages of clinical trials are tau transcription factor inhibitors (NCT04253132), tau aggregation inhibitors (AZP2006), and O-GlcNAcase inhibitors (hypothesized to help with tau hyperphosphorylation from transgenic mice studies).[97] There has been a recent interest in tau-targeted antisense oligonucleotides (ASOs) in 4R-tauopathies given an ASO-mediated 50% reduction in tau translation and prevention of neuronal loss in transgenic mouse models.[101] MAPTRx, a tau-targeting ASO, was found to be safe and showed a dose-dependent reduction in the total-tau CSF concentration in mild AD in a phase 1 randomized trial, and phase 2 of this drug is ongoing.[102] Encouraging results will potentially expand clinical trials for these agents in 4R-tauopathies.
Differential Diagnosis
The most common clinical presentation of CBD, CBS, can have varying underlying pathologies other than CBD itself. These underlying pathologies can be PSP, AD, PD, diffuse Lewy body disease, Pick disease, Creutzfeldt-Jakob disease, cerebrovascular disease, motor neuron disease, frontotemporal lobar degeneration (FTLD)-TDP, FTLD-tau, or FTLD-FUS.[37][103] Patients with CBD who have cognitive predominant symptoms are more likely to have comorbid underlying AGD, a medial temporal tauopathy, when compared to CBD with CBS (73% vs 35%).[104]
Prognosis
Results from a study analyzing the natural history and survival of patients with confirmed CBD showed a median survival time from symptom onset of 7.9 years with a standard deviation of 0.7 years and a range of 2.5 to 12.5 years. After the first clinic visit, median survival was 4.9 years with a standard deviation of 0.7 years and a range of 0.8 to 10 years. Frontal syndrome, early bilateral bradykinesia, or 2 parkinsonian symptoms (tremor, bradykinesia, and rigidity) predicted a shorter survival.[22]
Complications
Patients with CBD are likely to accumulate progressive disability with disturbances in speech and language, swallowing, cognition, behavior, gait and posture stability, oculovisual functions, sleep, and possibly autonomic symptoms, eg, urinary dysfunction and constipation.[71]
Deterrence and Patient Education
Patient education and deterrence in CBD focus on providing patients and caregivers with a clear understanding of the disease course, symptom management, and strategies to maintain quality of life. Given the progressive and incurable nature of CBD, education should emphasize realistic expectations regarding disease progression, including worsening motor dysfunction, cognitive decline, and communication difficulties. Patients and families should be informed about the importance of early planning for mobility aids, speech therapy, and assistance with daily activities to maintain independence for as long as possible.
While no proven preventive measures for CBD exist, lifestyle modifications such as regular physical activity, cognitive stimulation, and a healthy diet may contribute to overall brain health. Patients should be encouraged to engage in physical therapy to maintain mobility and prevent complications like falls and joint contractures. Occupational therapy can assist with adaptive strategies for daily activities, while speech therapy can help manage dysphagia and communication difficulties.
Caregiver support is crucial, and families should be educated on available resources, including support groups, palliative care, and respite services. Discussions about advance care planning, including legal and financial considerations, should be introduced early to ensure the patient's wishes are respected as the disease progresses. By fostering a proactive approach, education and deterrence efforts can help patients and caregivers navigate the challenges of CBD with greater confidence and preparedness.
Enhancing Healthcare Team Outcomes
Effective corticobasal degeneration management requires a multidisciplinary approach emphasizing interprofessional communication, care coordination, and patient-centered strategies. Advanced clinicians must leverage their diagnostic and management expertise to develop personalized treatment plans while remaining vigilant for overlapping conditions like progressive supranuclear palsy or Alzheimer disease. Open communication between team members—such as neurologists, neuropsychologists, physical and occupational therapists, and speech-language pathologists—is crucial to address CBD’s diverse motor, cognitive, and behavioral manifestations. Regular interdisciplinary case reviews and care conferences can facilitate information sharing, reduce diagnostic delays, and ensure unified treatment goals, ultimately enhancing outcomes and patient safety.
Care coordination is equally critical to streamline therapies and avoid fragmentation of care. Pharmacists can ensure safe, evidence-based medication management for symptoms such as rigidity or dystonia while educating patients and caregivers about potential side effects. Nurses play a pivotal role in symptom monitoring, patient education, and caregiver support while serving as liaisons among providers. Social workers and case managers address psychosocial challenges and ensure access to community resources. Employing team strategies like shared decision-making, clear goal-setting, and patient education enhances patient-centered care, improves quality of life, and fosters a collaborative, high-performing team dynamic in managing the complexities of CBD.
Media
(Click Image to Enlarge)
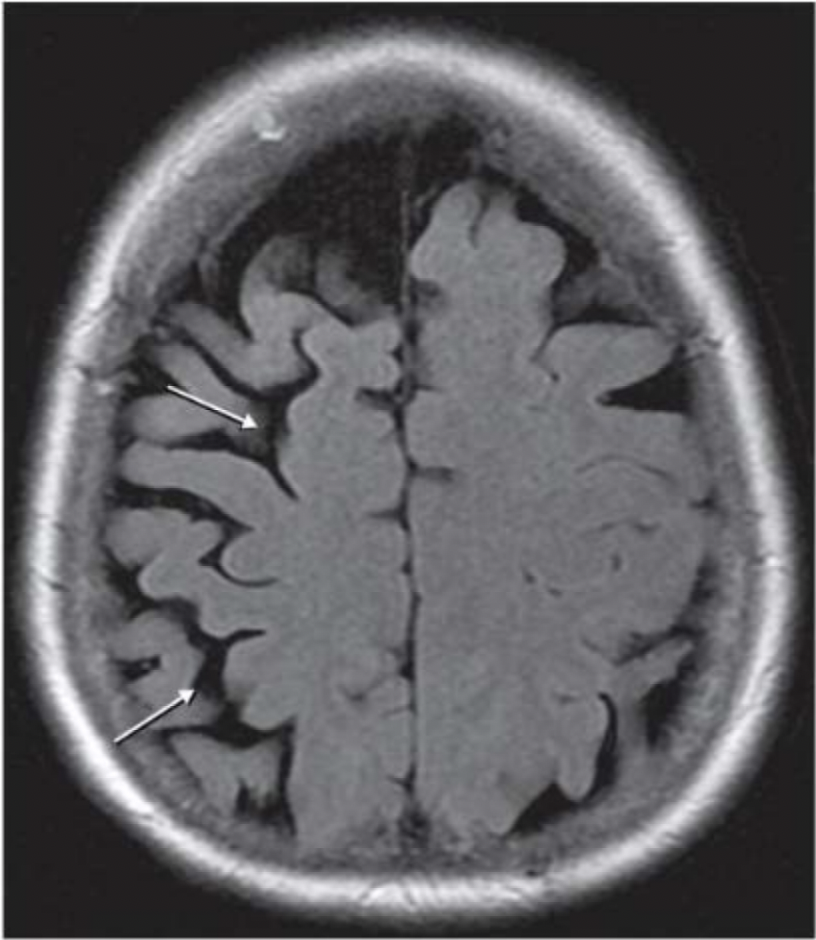
Corticobasal Degeneration on Magnetic Resonance Imaging. This is an axial T1 weighted magnetic resonance image showing asymmetric frontoparietal atrophy (white arrows) in a patient with corticobasal syndrome.
Chiu SY, Cutsforth-Gregory JK. Atypical Parkinsonian syndromes. In: Fleming KD, ed. Mayo Clinic Neurology Board Review. 2nd ed. New York, NY: Oxford University Press; 2022:chap 70.
(Click Image to Enlarge)
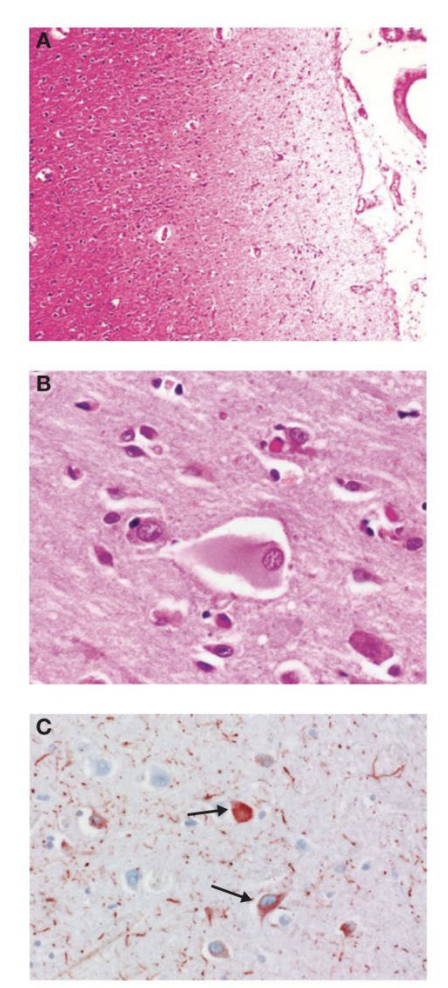
Histopathologic Characteristics of Corticobasal Degeneration. A) Superficial layers of the cerebral cortex demonstrating neuronal loss and microvacuolation are shown. B) Cortical neurons are characteristically swollen. C) Tau-positive intraneuronal inclusions appearing as globular or angular inclusions (arrows) are shown.
Chiu SY, Cutsforth-Gregory JK. Atypical Parkinsonian syndromes. In: Fleming KD, ed. Mayo Clinic Neurology Board Review. 2nd ed. New York, NY: Oxford University Press; 2022:chap 70.
References
Ali F, Josephs KA. Corticobasal degeneration: key emerging issues. Journal of neurology. 2018 Feb:265(2):439-445. doi: 10.1007/s00415-017-8644-3. Epub 2017 Oct 23 [PubMed PMID: 29063240]
Grijalvo-Perez AM, Litvan I. Corticobasal degeneration. Seminars in neurology. 2014 Apr:34(2):160-73. doi: 10.1055/s-0034-1381734. Epub 2014 Jun 25 [PubMed PMID: 24963675]
Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. Journal of neurology. 1999 Sep:246 Suppl 2():II6-15 [PubMed PMID: 10525997]
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. The Journal of biological chemistry. 1986 May 5:261(13):6084-9 [PubMed PMID: 3084478]
Brown J, Lantos PL, Roques P, Fidani L, Rossor MN. Familial dementia with swollen achromatic neurons and corticobasal inclusion bodies: a clinical and pathological study. Journal of the neurological sciences. 1996 Jan:135(1):21-30 [PubMed PMID: 8926492]
Saranza GM, Whitwell JL, Kovacs GG, Lang AE. Corticobasal degeneration. International review of neurobiology. 2019:149():87-136. doi: 10.1016/bs.irn.2019.10.014. Epub 2019 Nov 21 [PubMed PMID: 31779825]
Houlden H, Baker M, Morris HR, MacDonald N, Pickering-Brown S, Adamson J, Lees AJ, Rossor MN, Quinn NP, Kertesz A, Khan MN, Hardy J, Lantos PL, St George-Hyslop P, Munoz DG, Mann D, Lang AE, Bergeron C, Bigio EH, Litvan I, Bhatia KP, Dickson D, Wood NW, Hutton M. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001 Jun 26:56(12):1702-6 [PubMed PMID: 11425937]
Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, Baker M, Finch NCA, Yoon H, Kim J, Fujioka S, McLean CA, Ghetti B, Spina S, Cantwell LB, Farlow MR, Grafman J, Huey ED, Ryung Han M, Beecher S, Geller ET, Kretzschmar HA, Roeber S, Gearing M, Juncos JL, Vonsattel JPG, Van Deerlin VM, Grossman M, Hurtig HI, Gross RG, Arnold SE, Trojanowski JQ, Lee VM, Wenning GK, White CL, Höglinger GU, Müller U, Devlin B, Golbe LI, Crook J, Parisi JE, Boeve BF, Josephs KA, Wszolek ZK, Uitti RJ, Graff-Radford NR, Litvan I, Younkin SG, Wang LS, Ertekin-Taner N, Rademakers R, Hakonarsen H, Schellenberg GD, Dickson DW. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nature communications. 2015 Jun 16:6():7247. doi: 10.1038/ncomms8247. Epub 2015 Jun 16 [PubMed PMID: 26077951]
Robinson JL, Yan N, Caswell C, Xie SX, Suh E, Van Deerlin VM, Gibbons G, Irwin DJ, Grossman M, Lee EB, Lee VM, Miller B, Trojanowski JQ. Primary Tau Pathology, Not Copathology, Correlates With Clinical Symptoms in PSP and CBD. Journal of neuropathology and experimental neurology. 2020 Mar 1:79(3):296-304. doi: 10.1093/jnen/nlz141. Epub [PubMed PMID: 31999351]
Zhang CC, Zhu JX, Wan Y, Tan L, Wang HF, Yu JT, Tan L. Meta-analysis of the association between variants in MAPT and neurodegenerative diseases. Oncotarget. 2017 Jul 4:8(27):44994-45007. doi: 10.18632/oncotarget.16690. Epub [PubMed PMID: 28402959]
Level 1 (high-level) evidenceForrest SL, Kril JJ, Stevens CH, Kwok JB, Hallupp M, Kim WS, Huang Y, McGinley CV, Werka H, Kiernan MC, Götz J, Spillantini MG, Hodges JR, Ittner LM, Halliday GM. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain : a journal of neurology. 2018 Feb 1:141(2):521-534. doi: 10.1093/brain/awx328. Epub [PubMed PMID: 29253099]
Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathology and applied neurobiology. 2015 Feb:41(1):24-46. doi: 10.1111/nan.12213. Epub [PubMed PMID: 25556536]
Yokoyama JS, Karch CM, Fan CC, Bonham LW, Kouri N, Ross OA, Rademakers R, Kim J, Wang Y, Höglinger GU, Müller U, Ferrari R, Hardy J, International FTD-Genomics Consortium (IFGC), Momeni P, Sugrue LP, Hess CP, James Barkovich A, Boxer AL, Seeley WW, Rabinovici GD, Rosen HJ, Miller BL, Schmansky NJ, Fischl B, Hyman BT, Dickson DW, Schellenberg GD, Andreassen OA, Dale AM, Desikan RS. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta neuropathologica. 2017 May:133(5):825-837. doi: 10.1007/s00401-017-1693-y. Epub 2017 Mar 7 [PubMed PMID: 28271184]
Broce I, Karch CM, Wen N, Fan CC, Wang Y, Tan CH, Kouri N, Ross OA, Höglinger GU, Muller U, Hardy J, International FTD-Genomics Consortium, Momeni P, Hess CP, Dillon WP, Miller ZA, Bonham LW, Rabinovici GD, Rosen HJ, Schellenberg GD, Franke A, Karlsen TH, Veldink JH, Ferrari R, Yokoyama JS, Miller BL, Andreassen OA, Dale AM, Desikan RS, Sugrue LP. Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS medicine. 2018 Jan:15(1):e1002487. doi: 10.1371/journal.pmed.1002487. Epub 2018 Jan 9 [PubMed PMID: 29315334]
Fekete R, Bainbridge M, Baizabal-Carvallo JF, Rivera A, Miller B, Du P, Kholodovych V, Powell S, Ondo W. Exome sequencing in familial corticobasal degeneration. Parkinsonism & related disorders. 2013 Nov:19(11):1049-52. doi: 10.1016/j.parkreldis.2013.06.016. Epub 2013 Jul 16 [PubMed PMID: 23867865]
Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, Boxer AL, Dickson DW, Grossman M, Hallett M, Josephs KA, Kertesz A, Lee SE, Miller BL, Reich SG, Riley DE, Tolosa E, Tröster AI, Vidailhet M, Weiner WJ. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013 Jan 29:80(5):496-503. doi: 10.1212/WNL.0b013e31827f0fd1. Epub [PubMed PMID: 23359374]
Lyons S, Trépel D, Lynch T, Walsh R, O'Dowd S. The prevalence and incidence of progressive supranuclear palsy and corticobasal syndrome: a systematic review and meta-analysis. Journal of neurology. 2023 Sep:270(9):4451-4465. doi: 10.1007/s00415-023-11791-2. Epub 2023 Jun 8 [PubMed PMID: 37289323]
Level 1 (high-level) evidenceOsaki Y, Morita Y, Kuwahara T, Miyano I, Doi Y. Prevalence of Parkinson's disease and atypical parkinsonian syndromes in a rural Japanese district. Acta neurologica Scandinavica. 2011 Sep:124(3):182-7. doi: 10.1111/j.1600-0404.2010.01442.x. Epub 2010 Sep 29 [PubMed PMID: 20880268]
Winter Y, Bezdolnyy Y, Katunina E, Avakjan G, Reese JP, Klotsche J, Oertel WH, Dodel R, Gusev E. Incidence of Parkinson's disease and atypical parkinsonism: Russian population-based study. Movement disorders : official journal of the Movement Disorder Society. 2010 Feb 15:25(3):349-56. doi: 10.1002/mds.22966. Epub [PubMed PMID: 20108378]
Togasaki DM, Tanner CM. Epidemiologic aspects. Advances in neurology. 2000:82():53-9 [PubMed PMID: 10624470]
Level 3 (low-level) evidenceSchrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet (London, England). 1999 Nov 20:354(9192):1771-5 [PubMed PMID: 10577638]
Level 2 (mid-level) evidenceWenning GK, Litvan I, Jankovic J, Granata R, Mangone CA, McKee A, Poewe W, Jellinger K, Ray Chaudhuri K, D'Olhaberriague L, Pearce RK. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. Journal of neurology, neurosurgery, and psychiatry. 1998 Feb:64(2):184-9 [PubMed PMID: 9489528]
Constantinides VC, Paraskevas GP, Paraskevas PG, Stefanis L, Kapaki E. Corticobasal degeneration and corticobasal syndrome: A review. Clinical parkinsonism & related disorders. 2019:1():66-71. doi: 10.1016/j.prdoa.2019.08.005. Epub 2019 Aug 30 [PubMed PMID: 34316603]
Kompoliti K, Goetz CG, Boeve BF, Maraganore DM, Ahlskog JE, Marsden CD, Bhatia KP, Greene PE, Przedborski S, Seal EC, Burns RS, Hauser RA, Gauger LL, Factor SA, Molho ES, Riley DE. Clinical presentation and pharmacological therapy in corticobasal degeneration. Archives of neurology. 1998 Jul:55(7):957-61 [PubMed PMID: 9678313]
Murray R, Neumann M, Forman MS, Farmer J, Massimo L, Rice A, Miller BL, Johnson JK, Clark CM, Hurtig HI, Gorno-Tempini ML, Lee VM, Trojanowski JQ, Grossman M. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology. 2007 Apr 17:68(16):1274-83 [PubMed PMID: 17438218]
Witman GB, Cleveland DW, Weingarten MD, Kirschner MW. Tubulin requires tau for growth onto microtubule initiating sites. Proceedings of the National Academy of Sciences of the United States of America. 1976 Nov:73(11):4070-4 [PubMed PMID: 1069293]
Ayers JI, Giasson BI, Borchelt DR. Prion-like Spreading in Tauopathies. Biological psychiatry. 2018 Feb 15:83(4):337-346. doi: 10.1016/j.biopsych.2017.04.003. Epub 2017 Apr 13 [PubMed PMID: 28506438]
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010 Aug 6:142(3):387-97. doi: 10.1016/j.cell.2010.06.036. Epub 2010 Jul 22 [PubMed PMID: 20655099]
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010 Dec 22:68(6):1067-81. doi: 10.1016/j.neuron.2010.11.030. Epub [PubMed PMID: 21172610]
Ling H, Kovacs GG, Vonsattel JP, Davey K, Mok KY, Hardy J, Morris HR, Warner TT, Holton JL, Revesz T. Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain : a journal of neurology. 2016 Dec:139(Pt 12):3237-3252 [PubMed PMID: 27797812]
Boeve BF, Maraganore DM, Parisi JE, Ahlskog JE, Graff-Radford N, Caselli RJ, Dickson DW, Kokmen E, Petersen RC. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology. 1999 Sep 11:53(4):795-800 [PubMed PMID: 10489043]
Level 3 (low-level) evidenceIrwin DJ. Tauopathies as clinicopathological entities. Parkinsonism & related disorders. 2016 Jan:22 Suppl 1(0 1):S29-33. doi: 10.1016/j.parkreldis.2015.09.020. Epub 2015 Sep 8 [PubMed PMID: 26382841]
Querol-Vilaseca M, Colom-Cadena M, Pegueroles J, San Martín-Paniello C, Clarimon J, Belbin O, Fortea J, Lleó A. YKL-40 (Chitinase 3-like I) is expressed in a subset of astrocytes in Alzheimer's disease and other tauopathies. Journal of neuroinflammation. 2017 Jun 9:14(1):118. doi: 10.1186/s12974-017-0893-7. Epub 2017 Jun 9 [PubMed PMID: 28599675]
Ferrer I, López-González I, Carmona M, Arregui L, Dalfó E, Torrejón-Escribano B, Diehl R, Kovacs GG. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. Journal of neuropathology and experimental neurology. 2014 Jan:73(1):81-97. doi: 10.1097/NEN.0000000000000030. Epub [PubMed PMID: 24335532]
Murray ME, Kouri N, Lin WL, Jack CR Jr, Dickson DW, Vemuri P. Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alzheimer's research & therapy. 2014:6(1):1. doi: 10.1186/alzrt231. Epub 2014 Jan 2 [PubMed PMID: 24382028]
Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I, Office of Rare Diseases of the National Institutes of Health. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. Journal of neuropathology and experimental neurology. 2002 Nov:61(11):935-46 [PubMed PMID: 12430710]
Koga S, Josephs KA, Aiba I, Yoshida M, Dickson DW. Neuropathology and emerging biomarkers in corticobasal syndrome. Journal of neurology, neurosurgery, and psychiatry. 2022 Jun 13:93(9):919-29. doi: 10.1136/jnnp-2021-328586. Epub 2022 Jun 13 [PubMed PMID: 35697501]
Kouri N, Murray ME, Hassan A, Rademakers R, Uitti RJ, Boeve BF, Graff-Radford NR, Wszolek ZK, Litvan I, Josephs KA, Dickson DW. Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain : a journal of neurology. 2011 Nov:134(Pt 11):3264-75. doi: 10.1093/brain/awr234. Epub 2011 Sep 20 [PubMed PMID: 21933807]
Koga S, Kouri N, Walton RL, Ebbert MTW, Josephs KA, Litvan I, Graff-Radford N, Ahlskog JE, Uitti RJ, van Gerpen JA, Boeve BF, Parks A, Ross OA, Dickson DW. Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: a distinct clinicopathologic subtype. Acta neuropathologica. 2018 Sep:136(3):389-404. doi: 10.1007/s00401-018-1878-z. Epub 2018 Jun 20 [PubMed PMID: 29926172]
Ouchi H, Toyoshima Y, Tada M, Oyake M, Aida I, Tomita I, Satoh A, Tsujihata M, Takahashi H, Nishizawa M, Shimohata T. Pathology and sensitivity of current clinical criteria in corticobasal syndrome. Movement disorders : official journal of the Movement Disorder Society. 2014 Feb:29(2):238-44. doi: 10.1002/mds.25746. Epub 2013 Nov 20 [PubMed PMID: 24259271]
Hassan A, Whitwell JL, Boeve BF, Jack CR Jr, Parisi JE, Dickson DW, Josephs KA. Symmetric corticobasal degeneration (S-CBD). Parkinsonism & related disorders. 2010 Mar:16(3):208-14. doi: 10.1016/j.parkreldis.2009.11.013. Epub 2009 Dec 16 [PubMed PMID: 20018548]
Frucht S, Fahn S, Chin S, Dhawan V, Eidelberg D. Levodopa-induced dyskinesias in autopsy-proven cortical-basal ganglionic degeneration. Movement disorders : official journal of the Movement Disorder Society. 2000 Mar:15(2):340-3 [PubMed PMID: 10752591]
Müller J, Seppi K, Stefanova N, Poewe W, Litvan I, Wenning GK. Freezing of gait in postmortem-confirmed atypical parkinsonism. Movement disorders : official journal of the Movement Disorder Society. 2002 Sep:17(5):1041-5 [PubMed PMID: 12360556]
Factor SA, Higgins DS, Qian J. Primary progressive freezing gait: a syndrome with many causes. Neurology. 2006 Feb 14:66(3):411-4 [PubMed PMID: 16476942]
Level 2 (mid-level) evidenceStamelou M, Alonso-Canovas A, Bhatia KP. Dystonia in corticobasal degeneration: a review of the literature on 404 pathologically proven cases. Movement disorders : official journal of the Movement Disorder Society. 2012 May:27(6):696-702. doi: 10.1002/mds.24992. Epub 2012 May 1 [PubMed PMID: 22550031]
Level 3 (low-level) evidenceGodeiro-Junior C, Felício AC, Barsottini OG, Aguiar PM, Silva SM, Borges V, Ferraz HB. Clinical features of dystonia in atypical parkinsonism. Arquivos de neuro-psiquiatria. 2008 Dec:66(4):800-4 [PubMed PMID: 19099114]
Vanek ZF, Jankovic J. Dystonia in corticobasal degeneration. Advances in neurology. 2000:82():61-7 [PubMed PMID: 10624471]
Level 3 (low-level) evidenceChen R, Ashby P, Lang AE. Stimulus-sensitive myoclonus in akinetic-rigid syndromes. Brain : a journal of neurology. 1992 Dec:115 ( Pt 6)():1875-88 [PubMed PMID: 1486465]
Ling H, O'Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, Paviour DC, Lees AJ. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain : a journal of neurology. 2010 Jul:133(Pt 7):2045-57. doi: 10.1093/brain/awq123. Epub [PubMed PMID: 20584946]
Thompson PD, Day BL, Rothwell JC, Brown P, Britton TC, Marsden CD. The myoclonus in corticobasal degeneration. Evidence for two forms of cortical reflex myoclonus. Brain : a journal of neurology. 1994 Oct:117 ( Pt 5)():1197-207 [PubMed PMID: 7953599]
Thompson PD, Shibasaki H. Myoclonus in corticobasal degeneration and other neurodegenerations. Advances in neurology. 2000:82():69-81 [PubMed PMID: 10624472]
Level 3 (low-level) evidenceRinne JO, Lee MS, Thompson PD, Marsden CD. Corticobasal degeneration. A clinical study of 36 cases. Brain : a journal of neurology. 1994 Oct:117 ( Pt 5)():1183-96 [PubMed PMID: 7953598]
Level 3 (low-level) evidenceMüller J, Wenning GK, Verny M, McKee A, Chaudhuri KR, Jellinger K, Poewe W, Litvan I. Progression of dysarthria and dysphagia in postmortem-confirmed parkinsonian disorders. Archives of neurology. 2001 Feb:58(2):259-64 [PubMed PMID: 11176964]
Leiguarda R, Merello M, Balej J. Apraxia in corticobasal degeneration. Advances in neurology. 2000:82():103-21 [PubMed PMID: 10624475]
Level 3 (low-level) evidenceZadikoff C, Lang AE. Apraxia in movement disorders. Brain : a journal of neurology. 2005 Jul:128(Pt 7):1480-97 [PubMed PMID: 15930045]
Lang AE. Cortical basal ganglionic degeneration presenting with "progressive loss of speech output and orofacial dyspraxia". Journal of neurology, neurosurgery, and psychiatry. 1992 Nov:55(11):1101 [PubMed PMID: 1469415]
Huey ED, Pardini M, Cavanagh A, Wassermann EM, Kapogiannis D, Spina S, Ghetti B, Grafman J. Association of ideomotor apraxia with frontal gray matter volume loss in corticobasal syndrome. Archives of neurology. 2009 Oct:66(10):1274-80. doi: 10.1001/archneurol.2009.218. Epub [PubMed PMID: 19822784]
Frattali CM, Grafman J, Patronas N, Makhlouf F, Litvan I. Language disturbances in corticobasal degeneration. Neurology. 2000 Feb 22:54(4):990-2 [PubMed PMID: 10691002]
Grimes DA, Lang AE, Bergeron CB. Dementia as the most common presentation of cortical-basal ganglionic degeneration. Neurology. 1999 Dec 10:53(9):1969-74 [PubMed PMID: 10599767]
Levin J, Bak TH, Rominger A, Mille E, Arzberger T, Giese A, Ackl N, Lorenzl S, Bader B, Patzig M, Bötzel K, Danek A. The association of aphasia and right-sided motor impairment in corticobasal syndrome. Journal of neurology. 2015 Oct:262(10):2241-6. doi: 10.1007/s00415-015-7833-1. Epub 2015 Jul 5 [PubMed PMID: 26143172]
Brion S, Jedynak CP. [Disorders of interhemispheric transfer (callosal disonnection). 3 cases of tumor of the corpus callosum. The strange hand sign]. Revue neurologique. 1972 Apr:126(4):257-66 [PubMed PMID: 4350533]
Level 3 (low-level) evidenceDoody RS, Jankovic J. The alien hand and related signs. Journal of neurology, neurosurgery, and psychiatry. 1992 Sep:55(9):806-10 [PubMed PMID: 1402972]
Bundick T Jr, Spinella M. Subjective experience, involuntary movement, and posterior alien hand syndrome. Journal of neurology, neurosurgery, and psychiatry. 2000 Jan:68(1):83-5 [PubMed PMID: 10601408]
Biran I, Chatterjee A. Alien hand syndrome. Archives of neurology. 2004 Feb:61(2):292-4 [PubMed PMID: 14967782]
DENNY-BROWN D. The nature of apraxia. The Journal of nervous and mental disease. 1958 Jan:126(1):9-32 [PubMed PMID: 13514485]
Fitzgerald DB, Drago V, Jeong Y, Chang YL, White KD, Heilman KM. Asymmetrical alien hands in corticobasal degeneration. Movement disorders : official journal of the Movement Disorder Society. 2007 Mar 15:22(4):581-4 [PubMed PMID: 17230447]
Bergeron C, Davis A, Lang AE. Corticobasal ganglionic degeneration and progressive supranuclear palsy presenting with cognitive decline. Brain pathology (Zurich, Switzerland). 1998 Apr:8(2):355-65 [PubMed PMID: 9546292]
Kimura N, Kumamoto T, Hanaoka T, Hazama Y, Nakamura K, Arakawa R. Corticobasal degeneration presenting with progressive conduction aphasia. Journal of the neurological sciences. 2008 Jun 15:269(1-2):163-8. doi: 10.1016/j.jns.2007.12.017. Epub 2008 Jan 29 [PubMed PMID: 18230400]
Rajagopal R, Bateman R, Van Stavern GP. Visual involvement in corticobasal syndrome. Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society. 2012 Dec:32(4):338-40. doi: 10.1097/WNO.0b013e3182305162. Epub [PubMed PMID: 21926916]
Alster P, Madetko-Alster N, Migda A, Migda B, Kutyłowski M, Królicki L, Friedman A. Sleep disturbances in progressive supranuclear palsy syndrome (PSPS) and corticobasal syndrome (CBS). Neurologia i neurochirurgia polska. 2023:57(3):229-234. doi: 10.5603/PJNNS.a2023.0019. Epub 2023 Mar 17 [PubMed PMID: 36928793]
Bluett B, Pantelyat AY, Litvan I, Ali F, Apetauerova D, Bega D, Bloom L, Bower J, Boxer AL, Dale ML, Dhall R, Duquette A, Fernandez HH, Fleisher JE, Grossman M, Howell M, Kerwin DR, Leegwater-Kim J, Lepage C, Ljubenkov PA, Mancini M, McFarland NR, Moretti P, Myrick E, Patel P, Plummer LS, Rodriguez-Porcel F, Rojas J, Sidiropoulos C, Sklerov M, Sokol LL, Tuite PJ, VandeVrede L, Wilhelm J, Wills AA, Xie T, Golbe LI. Best Practices in the Clinical Management of Progressive Supranuclear Palsy and Corticobasal Syndrome: A Consensus Statement of the CurePSP Centers of Care. Frontiers in neurology. 2021:12():694872. doi: 10.3389/fneur.2021.694872. Epub 2021 Jul 1 [PubMed PMID: 34276544]
Level 3 (low-level) evidenceBruns MB, Josephs KA. Neuropsychiatry of corticobasal degeneration and progressive supranuclear palsy. International review of psychiatry (Abingdon, England). 2013 Apr:25(2):197-209. doi: 10.3109/09540261.2013.766154. Epub [PubMed PMID: 23611349]
Koga S, Aiba I. [Autonomic Dysfunction in Tauopathies]. Brain and nerve = Shinkei kenkyu no shinpo. 2022 Mar:74(3):257-262. doi: 10.11477/mf.1416202021. Epub [PubMed PMID: 35260524]
Armstrong RA. Oculo-Visual Dysfunction in Parkinson's Disease. Journal of Parkinson's disease. 2015:5(4):715-26. doi: 10.3233/JPD-150686. Epub [PubMed PMID: 26599301]
Rossor MN, Tyrrell PJ, Warrington EK, Thompson PD, Marsden CD, Lantos P. Progressive frontal gait disturbance with atypical Alzheimer's disease and corticobasal degeneration. Journal of neurology, neurosurgery, and psychiatry. 1999 Sep:67(3):345-52 [PubMed PMID: 10449557]
Horie K, Barthélemy NR, Spina S, VandeVrede L, He Y, Paterson RW, Wright BA, Day GS, Davis AA, Karch CM, Seeley WW, Perrin RJ, Koppisetti RK, Shaikh F, Lago AL, Heuer HW, Ghoshal N, Gabelle A, Miller BL, Boxer AL, Bateman RJ, Sato C. CSF tau microtubule-binding region identifies pathological changes in primary tauopathies. Nature medicine. 2022 Dec:28(12):2547-2554. doi: 10.1038/s41591-022-02075-9. Epub 2022 Nov 24 [PubMed PMID: 36424467]
Mattsson N, Insel PS, Donohue M, Jagust W, Sperling R, Aisen P, Weiner MW, Alzheimer’s Disease Neuroimaging Initiative. Predicting Reduction of Cerebrospinal Fluid β-Amyloid 42 in Cognitively Healthy Controls. JAMA neurology. 2015 May:72(5):554-60. doi: 10.1001/jamaneurol.2014.4530. Epub [PubMed PMID: 25775167]
Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, Huang EJ, Trojanowski JQ, Growdon ME, Jang JY, Sidhu M, See TM, Karydas AM, Gorno-Tempini ML, Boxer AL, Weiner MW, Geschwind MD, Rankin KP, Miller BL. Clinicopathological correlations in corticobasal degeneration. Annals of neurology. 2011 Aug:70(2):327-40. doi: 10.1002/ana.22424. Epub [PubMed PMID: 21823158]
Whitwell JL, Jack CR Jr, Boeve BF, Parisi JE, Ahlskog JE, Drubach DA, Senjem ML, Knopman DS, Petersen RC, Dickson DW, Josephs KA. Imaging correlates of pathology in corticobasal syndrome. Neurology. 2010 Nov 23:75(21):1879-87. doi: 10.1212/WNL.0b013e3181feb2e8. Epub [PubMed PMID: 21098403]
Carlos AF, Tosakulwong N, Weigand SD, Buciuc M, Ali F, Clark HM, Botha H, Utianski RL, Machulda MM, Schwarz CG, Reid RI, Senjem ML, Jack CR Jr, Ahlskog JE, Dickson DW, Josephs KA, Whitwell JL. Histologic lesion type correlates of magnetic resonance imaging biomarkers in four-repeat tauopathies. Brain communications. 2022:4(3):fcac108. doi: 10.1093/braincomms/fcac108. Epub 2022 Apr 28 [PubMed PMID: 35663380]
Erbetta A, Mandelli ML, Savoiardo M, Grisoli M, Bizzi A, Soliveri P, Chiapparini L, Prioni S, Bruzzone MG, Girotti F. Diffusion tensor imaging shows different topographic involvement of the thalamus in progressive supranuclear palsy and corticobasal degeneration. AJNR. American journal of neuroradiology. 2009 Sep:30(8):1482-7. doi: 10.3174/ajnr.A1615. Epub 2009 Jul 9 [PubMed PMID: 19589886]
Teune LK, Bartels AL, de Jong BM, Willemsen AT, Eshuis SA, de Vries JJ, van Oostrom JC, Leenders KL. Typical cerebral metabolic patterns in neurodegenerative brain diseases. Movement disorders : official journal of the Movement Disorder Society. 2010 Oct 30:25(14):2395-404. doi: 10.1002/mds.23291. Epub [PubMed PMID: 20669302]
Niethammer M, Tang CC, Feigin A, Allen PJ, Heinen L, Hellwig S, Amtage F, Hanspal E, Vonsattel JP, Poston KL, Meyer PT, Leenders KL, Eidelberg D. A disease-specific metabolic brain network associated with corticobasal degeneration. Brain : a journal of neurology. 2014 Nov:137(Pt 11):3036-46. doi: 10.1093/brain/awu256. Epub 2014 Sep 9 [PubMed PMID: 25208922]
Pardini M, Huey ED, Spina S, Kreisl WC, Morbelli S, Wassermann EM, Nobili F, Ghetti B, Grafman J. FDG-PET patterns associated with underlying pathology in corticobasal syndrome. Neurology. 2019 Mar 5:92(10):e1121-e1135. doi: 10.1212/WNL.0000000000007038. Epub 2019 Jan 30 [PubMed PMID: 30700592]
Mena AM, Strafella AP. Imaging pathological tau in atypical parkinsonisms: A review. Clinical parkinsonism & related disorders. 2022:7():100155. doi: 10.1016/j.prdoa.2022.100155. Epub 2022 Jul 16 [PubMed PMID: 35880206]
Murugan NA, Nordberg A, Ågren H. Cryptic Sites in Tau Fibrils Explain the Preferential Binding of the AV-1451 PET Tracer toward Alzheimer's Tauopathy. ACS chemical neuroscience. 2021 Jul 7:12(13):2437-2447. doi: 10.1021/acschemneuro.0c00340. Epub 2021 Jun 21 [PubMed PMID: 34152739]
Murugan NA, Chiotis K, Rodriguez-Vieitez E, Lemoine L, Ågren H, Nordberg A. Cross-interaction of tau PET tracers with monoamine oxidase B: evidence from in silico modelling and in vivo imaging. European journal of nuclear medicine and molecular imaging. 2019 Jun:46(6):1369-1382. doi: 10.1007/s00259-019-04305-8. Epub 2019 Mar 27 [PubMed PMID: 30919054]
Soleimani-Meigooni DN, Iaccarino L, La Joie R, Baker S, Bourakova V, Boxer AL, Edwards L, Eser R, Gorno-Tempini ML, Jagust WJ, Janabi M, Kramer JH, Lesman-Segev OH, Mellinger T, Miller BL, Pham J, Rosen HJ, Spina S, Seeley WW, Strom A, Grinberg LT, Rabinovici GD. 18F-flortaucipir PET to autopsy comparisons in Alzheimer's disease and other neurodegenerative diseases. Brain : a journal of neurology. 2020 Dec 5:143(11):3477-3494. doi: 10.1093/brain/awaa276. Epub [PubMed PMID: 33141172]
Zhou XY, Lu JY, Liu FT, Wu P, Zhao J, Ju ZZ, Tang YL, Shi QY, Lin HM, Wu JJ, Yen TC, Zuo CT, Sun YM, Wang J. In Vivo (18) F-APN-1607 Tau Positron Emission Tomography Imaging in MAPT Mutations: Cross-Sectional and Longitudinal Findings. Movement disorders : official journal of the Movement Disorder Society. 2022 Mar:37(3):525-534. doi: 10.1002/mds.28867. Epub 2021 Nov 29 [PubMed PMID: 34842301]
Level 2 (mid-level) evidenceMashima K, Konishi M, Tezuka T, Ito D, Mimura M. A case of tauopathy with auditory agnosia and dysprosody diagnosed by [(18)F]PM-PBB3 tau PET scan. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2021 Aug:42(8):3471-3474. doi: 10.1007/s10072-021-05287-y. Epub 2021 May 4 [PubMed PMID: 33945032]
Level 3 (low-level) evidenceOh M, Oh SJ, Lee SJ, Oh JS, Roh JH, Chung SJ, Lee JH, Lee CS, Kim JS. Clinical Evaluation of 18F-PI-2620 as a Potent PET Radiotracer Imaging Tau Protein in Alzheimer Disease and Other Neurodegenerative Diseases Compared With 18F-THK-5351. Clinical nuclear medicine. 2020 Nov:45(11):841-847. doi: 10.1097/RLU.0000000000003261. Epub [PubMed PMID: 32910050]
Palleis C, Brendel M, Finze A, Weidinger E, Bötzel K, Danek A, Beyer L, Nitschmann A, Kern M, Biechele G, Rauchmann BS, Häckert J, Höllerhage M, Stephens AW, Drzezga A, van Eimeren T, Villemagne VL, Schildan A, Barthel H, Patt M, Sabri O, German Imaging Initiative for Tauopathies (GII4T), Bartenstein P, Perneczky R, Haass C, Levin J, Höglinger GU. Cortical [(18) F]PI-2620 Binding Differentiates Corticobasal Syndrome Subtypes. Movement disorders : official journal of the Movement Disorder Society. 2021 Sep:36(9):2104-2115. doi: 10.1002/mds.28624. Epub 2021 May 5 [PubMed PMID: 33951244]
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, Froelich L, Katayama S, Sabbagh M, Vellas B, Watson D, Dhadda S, Irizarry M, Kramer LD, Iwatsubo T. Lecanemab in Early Alzheimer's Disease. The New England journal of medicine. 2023 Jan 5:388(1):9-21. doi: 10.1056/NEJMoa2212948. Epub 2022 Nov 29 [PubMed PMID: 36449413]
Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, Wessels AM, Shcherbinin S, Wang H, Monkul Nery ES, Collins EC, Solomon P, Salloway S, Apostolova LG, Hansson O, Ritchie C, Brooks DA, Mintun M, Skovronsky DM, TRAILBLAZER-ALZ 2 Investigators. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 2023 Aug 8:330(6):512-527. doi: 10.1001/jama.2023.13239. Epub [PubMed PMID: 37459141]
Level 1 (high-level) evidenceDam T, Boxer AL, Golbe LI, Höglinger GU, Morris HR, Litvan I, Lang AE, Corvol JC, Aiba I, Grundman M, Yang L, Tidemann-Miller B, Kupferman J, Harper K, Kamisoglu K, Wald MJ, Graham DL, Gedney L, O'Gorman J, Haeberlein SB, PASSPORT Study Group. Safety and efficacy of anti-tau monoclonal antibody gosuranemab in progressive supranuclear palsy: a phase 2, randomized, placebo-controlled trial. Nature medicine. 2021 Aug:27(8):1451-1457. doi: 10.1038/s41591-021-01455-x. Epub 2021 Aug 12 [PubMed PMID: 34385707]
Khanna MR, Kovalevich J, Lee VM, Trojanowski JQ, Brunden KR. Therapeutic strategies for the treatment of tauopathies: Hopes and challenges. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2016 Oct:12(10):1051-1065. doi: 10.1016/j.jalz.2016.06.006. Epub [PubMed PMID: 27751442]
VandeVrede L, Ljubenkov PA, Rojas JC, Welch AE, Boxer AL. Four-Repeat Tauopathies: Current Management and Future Treatments. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2020 Oct:17(4):1563-1581. doi: 10.1007/s13311-020-00888-5. Epub [PubMed PMID: 32676851]
Bright J, Hussain S, Dang V, Wright S, Cooper B, Byun T, Ramos C, Singh A, Parry G, Stagliano N, Griswold-Prenner I. Human secreted tau increases amyloid-beta production. Neurobiology of aging. 2015 Feb:36(2):693-709. doi: 10.1016/j.neurobiolaging.2014.09.007. Epub 2014 Sep 16 [PubMed PMID: 25442111]
Courade JP, Angers R, Mairet-Coello G, Pacico N, Tyson K, Lightwood D, Munro R, McMillan D, Griffin R, Baker T, Starkie D, Nan R, Westwood M, Mushikiwabo ML, Jung S, Odede G, Sweeney B, Popplewell A, Burgess G, Downey P, Citron M. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta neuropathologica. 2018 Nov:136(5):729-745. doi: 10.1007/s00401-018-1911-2. Epub 2018 Sep 20 [PubMed PMID: 30238240]
Tsai RM, Miller Z, Koestler M, Rojas JC, Ljubenkov PA, Rosen HJ, Rabinovici GD, Fagan AM, Cobigo Y, Brown JA, Jung JI, Hare E, Geldmacher DS, Natelson-Love M, McKinley EC, Luong PN, Chuu EL, Powers R, Mumford P, Wolf A, Wang P, Shamloo M, Miller BL, Roberson ED, Boxer AL. Reactions to Multiple Ascending Doses of the Microtubule Stabilizer TPI-287 in Patients With Alzheimer Disease, Progressive Supranuclear Palsy, and Corticobasal Syndrome: A Randomized Clinical Trial. JAMA neurology. 2020 Feb 1:77(2):215-224. doi: 10.1001/jamaneurol.2019.3812. Epub [PubMed PMID: 31710340]
Level 1 (high-level) evidenceDeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B, Zanardi TA, Kordasiewicz HB, Swayze EE, Bennett CF, Diamond MI, Miller TM. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Science translational medicine. 2017 Jan 25:9(374):. doi: 10.1126/scitranslmed.aag0481. Epub [PubMed PMID: 28123067]
Mummery CJ, Börjesson-Hanson A, Blackburn DJ, Vijverberg EGB, De Deyn PP, Ducharme S, Jonsson M, Schneider A, Rinne JO, Ludolph AC, Bodenschatz R, Kordasiewicz H, Swayze EE, Fitzsimmons B, Mignon L, Moore KM, Yun C, Baumann T, Li D, Norris DA, Crean R, Graham DL, Huang E, Ratti E, Bennett CF, Junge C, Lane RM. Tau-targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer's disease: a phase 1b, randomized, placebo-controlled trial. Nature medicine. 2023 Jun:29(6):1437-1447. doi: 10.1038/s41591-023-02326-3. Epub 2023 Apr 24 [PubMed PMID: 37095250]
Shir D, Pham NTT, Botha H, Koga S, Kouri N, Ali F, Knopman DS, Petersen RC, Boeve BF, Kremers WK, Nguyen AT, Murray ME, Reichard RR, Dickson DW, Graff-Radford N, Josephs KA, Whitwell J, Graff-Radford J. Clinicoradiologic and Neuropathologic Evaluation of Corticobasal Syndrome. Neurology. 2023 Jul 18:101(3):e289-e299. doi: 10.1212/WNL.0000000000207397. Epub 2023 Jun 2 [PubMed PMID: 37268436]
Sakae N, Santos OA, Pedraza O, Litvan I, Murray ME, Duara R, Uitti RJ, Wszolek ZK, Graff-Radford NR, Josephs KA, Dickson DW. Clinical and pathologic features of cognitive-predominant corticobasal degeneration. Neurology. 2020 Jul 7:95(1):e35-e45. doi: 10.1212/WNL.0000000000009734. Epub 2020 Jun 9 [PubMed PMID: 32518146]