Introduction
Gastrointestinal neuroendocrine tumors (GI-NETs) are a diverse group of neoplasms that arise from neuroendocrine cells throughout the digestive tract. While these tumors can develop in various locations, the midgut is the most common site, particularly in the ileum and appendix. GI-NETs typically grow slowly; however, their clinical behavior is influenced by several factors, including differentiation status, histological grade, and the Ki-67 proliferation index. Poorly differentiated tumors, known as neuroendocrine carcinomas, are associated with more aggressive disease progression and poorer outcomes.[1]
Some GI-NETs are functional, producing bioactive hormones or peptides that can lead to specific clinical syndromes. Depending on their origin, these tumors can cause local symptoms, eg, intestinal obstruction, acute appendicitis, or bleeding. Metastases commonly occur in regional lymph nodes and the liver, leading to additional symptoms. Carcinoid syndrome, characterized by systemic symptoms due to circulating tumor products, may occur with midgut tumors that have significant liver metastases.[2]
Accurate diagnosis and staging of GI-NETs rely on cross-sectional imaging and, when appropriate, biochemical testing for hormone hypersecretion. Surgical resection is the primary treatment approach, especially when complete or near-complete removal of the tumor is possible. In situations where surgery is not feasible, other strategies, including medical management with somatostatin analogs, targeted therapies, or peptide receptor radionuclide therapy, are used for symptom relief and disease control.[2] This discussion focuses specifically on GI-NETs originating in the midgut and hindgut, while foregut and thoracic NETs are considered separately.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of GI-NETs is multifactorial and involves a combination of genetic, molecular, and environmental factors. Most GI-NETs are sporadic, and risk factors are not well understood. Approximately 20% of GI-NETs are inherited, such as multiple endocrine neoplasia type 1 (MEN1), von Hippel-Lindau disease (VHL), and neurofibromatosis (NF-1), and are associated with specific genetic mutations. Inherited GI-NETs are more commonly found in the foregut but are less likely to be responsible for midgut and hindgut GI-NETs.
Risk factors associated with GI-NETs include a positive family history of cancer, while lifestyle and metabolic factors may contribute to the development of these tumors. Obesity and diabetes have been implicated as potential risk factors, possibly due to their association with chronic inflammation and metabolic dysregulation. Some evidence has also suggested that cigarette smoking and alcohol use may play a role in increasing the risk of GI-NETs.[3] However, the exact mechanisms remain unclear, and more recent data suggest that several germline mutations may be responsible, at least in part, for some NETs.[4] These risk factors, genetic predispositions, and environmental influences highlight the multifactorial nature of GI-NET pathogenesis.[3]
Epidemiology
GI-NETs are less common than adenocarcinomas, yet their incidence has steadily increased.[1] GI-NETs represent the most common subtype of NETs, accounting for approximately 60% to 70% of cases, with the small intestine and appendix being the most frequently affected sites.[1] The annual incidence of GI-NETs is estimated to be 3 to 5 cases per 100,000 population, with a higher prevalence observed due to their typically indolent nature.[3] The incidence is also greater in more developed countries, attributable to greater availability of diagnostic testing and increased awareness. However, data suggests that the incidence also increases in less developed countries.[5]
These tumors are most commonly diagnosed in individuals aged between 50 and 70 and show a slight predominance in men. Non-Hispanic Black populations have higher incidence rates and worse survival outcomes than non-Hispanic White populations. However, the reasons for this disparity remain unclear. While most GI-NETs occur sporadically, approximately 20% are associated with hereditary syndromes (eg, MEN1, VHL, NF-1).[1] About 30% of GI-NETs are functional, producing bioactive hormones or peptides causing specific clinical syndromes, while the remaining 70% are nonfunctional.
Pathophysiology
GI-NETs arise from enterochromaffin and other neuroendocrine cells within the GI tract, which regulate hormones and peptides. The pathophysiology involves a combination of genetic mutations, epigenetic alterations, and dysregulated signaling pathways that promote unchecked cellular proliferation, survival, and hormonal secretion. Key gene mutations (eg, MEN1, DAXX, ATRX) and components of the mammalian target of rapamycin pathway disrupt normal cell cycle control and apoptosis. These tumors can produce excessive amounts of bioactive substances like serotonin, gastrin, or vasoactive intestinal peptide, leading to clinical syndromes (eg, carcinoid syndrome) when functional.[4][6]
Nonfunctional GI-NETs, while not associated with overt hormonal symptoms, may still grow locally invasive or metastasize. Differentiation, histological grade, and the Kiel-67 (Ki-67) proliferation index determine tumor behavior, including growth rate and metastatic potential. The angiogenic and stromal microenvironment also plays a critical role in tumor progression, with vascular endothelial growth factor and other proangiogenic signals contributing to the formation of the rich blood supply typical of these tumors.[4][6]
Histopathology
Neuroendocrine neoplasms (NENs) are classified based on degree of differentiation and grade. They are divided into well-differentiated NETs and poorly differentiated neuroendocrine carcinomas (NEC). GI-NETs are usually small, polyploid, solid, slow-growing, often invading transmurally, and spread to the lymphatics and adjacent mesentery. Jejunoileal NETs are frequently multifocal, with multiple lesions identified in up to 50% of cases.[6] NETs display monomorphic round cells with abundant eosinophilic cytoplasm and salt and pepper chromatin with rare mitotic figures. The internal structure may be solid, glandular, trabecular, or mixed, characterized by a highly vascular stroma with a low nuclear/cytoplasm ratio. Immunohistochemistry is positive for functional tumors' cytokeratins, chromogranin A, synaptophysin, somatostatin receptors, and various hormonal receptors.[6]
In contrast, neuroendocrine carcinoma (NEC) is characterized by polymorphic cells with frequent mitoses, necrosis, a high nuclear-to-cytoplasm ratio, and markedly aberrant cells. The degree of cytoplasm differentiates small and large cell types. The 2022 World Health Organization (WHO) classification of NETs based on tumor mitoses, Ki67 percent, and morphology is the most widely used (see Table. World Health Organization 2022 Classification of Neuroendocrine Tumors).[6]
Table. World Health Organization (WHO) 2022 Classification of Neuroendocrine Tumors
| Neuroendocrine Neoplasm | Classification | Diagnostic Criteria |
| Well-differentiated NET | NET, grade 1 | <2 mitoses/2 mm2or Ki67 <3% |
| NET, grade 2 | 2 to 20 mitoses/2 mm2 or Ki-67 3% to 20% | |
| NET, grade 3 | >20 mitoses/2 mm2 or Ki-67 >20% | |
| Poorly differentiated NEC | Small cell NEC | >20 mitoses/2 mm2or Ki-67 >20% (often >70%) and small cell cytomorphology |
| Large cell NEC | >20 mitoses/2 mm2 or Ki-67 >20% (often >70%) and large cell cytomorphology |
NEC, neuroendocrine carcinoma; NET, neuroendocrine tumors; Ki-67, Keil-67
History and Physical
The history and physical examination for GI-NETs focuses on identifying any association with genetic syndromes and symptoms associated with hormone hypersecretion, tumor mass effects, and potential complications. A history should encompass the evaluation of hallmark symptoms, including episodic flushing, watery diarrhea, and wheezing, which are characteristic of carcinoid syndrome, particularly in cases of metastatic disease associated with serotonin and other bioactive amine overproduction. Nonspecific manifestations include chronic abdominal pain, unintentional weight loss, early satiety, fatigue, and GI bleeding, which may indicate tumor burden, local invasion, or secondary complications. The regional lymph nodes and the liver are the most common sites of metastasis, with the bone, lung, peritoneum, and other organs being less common. A comprehensive review of systems is critical to detect subtle clinical features and to assess for predisposing conditions, eg, multiple endocrine neoplasia syndromes or von Hippel-Lindau syndrome.[7][8][9]
Physical examination findings often correlate with the tumor’s size, location, and extent of metastatic spread. Hepatomegaly, frequently indicative of metastatic liver involvement, should be carefully assessed through abdominal palpation. A palpable abdominal mass may suggest a primary tumor or significant regional lymphadenopathy. Signs of intestinal obstruction, such as abdominal distension, tenderness, or peritoneal irritation, may reflect tumor-related complications. Cutaneous findings, including telangiectasias, flushing, or pellagra-like dermatitis, may be observed in advanced disease due to niacin deficiency resulting from tryptophan diversion to serotonin synthesis. Furthermore, a thorough cardiovascular and respiratory evaluation is essential to identify features of carcinoid heart disease, eg, right-sided valvular dysfunction and associated murmurs.[7][8]
Evaluation
The diagnosis of GI-NETs is frequently delayed, often by 5 to 7 years for NETs, as patients are frequently asymptomatic or present with nonspecific symptoms. At the time of diagnosis, metastases are present in approximately 30% of those with NET, and this rate can reach as high as 70% in cases of midgut NETs. NECs, due to their aggressive nature, often present with advanced disease at diagnosis.
Biochemical Testing
Comprehensive biochemical testing is critical for the diagnosis and management of NETs. Chromogranin A (CgA) and a 24-hour urine collection for 5-hydroxyindoleacetic acid (5-HIAA) are useful, along with other testing based on clinical presentation.
- Chromogranin A Testing
- CgA is a nonspecific biomarker for NETs and is helpful for screening and monitoring of functioning and nonfunctioning NETs. CgA levels should be tested with the patient fasting and avoiding physical exertion. CgA levels correlate with tumor burden and may have prognostic implications. However, small tumors may produce normal CgA levels, while false-positive elevations can occur in the presence of severe hypertension, chronic gastritis, proton pump inhibitor use, and renal insufficiency. Given this lack of sensitivity and specificity, its utility is limited.[10][11][12]
- Twenty-Four-Hour Urine Collection
- Measuring 5-HIAA, a serotonin metabolite, with a 24-hour urine collection is particularly useful for diagnosing and monitoring serotonin-secreting GI-NETs, eg, midgut NETs. Before and during urine collection, patients should avoid serotonin and tryptophan-rich foods and medications that can lead to false elevations. Elevated levels in the setting of appropriate dietary and medication restrictions are highly suggestive of carcinoid syndrome.
- Serum serotonin measurements are not reliable due to variability related to daily activities. Although fasting plasma 5-HIAA testing offers convenience, additional validation is needed before it can replace the traditional 24-hour urine method. In NECs, specific biochemical markers depend on the functional status of the tumor.[13]
Genetic Assays
Genetic testing is essential for diagnosing hereditary GI-NETs. Gene expression tests are increasingly used in many cancers, which applies to NETs. An example is the NETest, a type of liquid biopsy that measures specific gene expression patterns in blood samples and generates a score indicating disease activity. Study results have shown that the NETest can achieve higher accuracy than standard biochemical tests (eg, CgA), with reported sensitivities of around 93% and specificities of about 98%. However, further research is needed before these tools become widely adopted in routine clinical practice.[14][15]
Tumor Circulation Markers
Markers of tumor circulation, including circulating tumor deoxyribonucleic acid and circulating tumor cells, are well-established in managing malignancies such as colorectal and breast cancer. Emerging evidence indicates these markers may also have clinical utility in NETs.[16]
Imaging Studies
Along with biochemical testing, a GI-NET diagnosis relies on imaging modalities, including computed tomography (CT), magnetic resonance imaging (MRI), and somatostatin receptor-based imaging.
- CT
- CT constitutes a critical component of the initial diagnostic workup for patients presenting with suspected GI-NETs, partly due to its widespread availability and rapid image acquisition. Most NETs demonstrate hyperenhancement on contrast-enhanced CT, particularly during arterial and venous phases (see Image. Neuroendocrine Tumor Liver Metastases). This makes a triple-phase protocol (noncontrast, arterial, and venous imaging) advantageous for lesion detection and staging. Reported sensitivities for CT in diagnosing NETs vary by tumor site and size, with estimates ranging from 60% to 85% in detecting small primary lesions. While CT is an effective modality for identifying hepatic metastases—often revealing hypervascular lesions on the arterial phase—CT is generally less sensitive than MRI, particularly for smaller (<1 cm) liver metastases.[17][18]
- MRI
- Some study results indicate that MRI can detect up to 90% of liver metastases from NETs, underscoring the complementary role of MRI in diagnostic algorithms for patients with suspected metastatic disease.[17][18] MRI is often more sensitive than CT for identifying hepatic metastases, which characteristically appear hypervascular on arterial-phase images and may show rapid washout in later phases. T1-weighted, T2-weighted, and diffusion-weighted sequences complement contrast-enhanced imaging by highlighting tumor cellularity and vascularity, thereby refining lesion detection and characterization.[17][19]
- Somatostatin receptor imaging
- Somatostatin receptor scintigraphy (eg, Octreoscan) and positron emission tomography (PET) imaging with DOTATOC (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid Tyr³-octreotide) or similar radiolabeled somatostatin analogs are highly effective for identifying somatostatin receptor-expressing tumors and evaluating the extent of disease. Please see StatPearls' companion resource, "Octreotide Scan," for further information. In NECs, these modalities may have limited utility due to poor differentiation and variable receptor expression.[18][20]
- Fluorodeoxyglucose positron emission tomography
Endoscopy
Upper and lower GI endoscopy allows for direct visualization and biopsy of accessible lesions. Enteroscopy or capsule endoscopy ("pill-cam") may be employed for small bowel lesions. However, routine use of capsule endoscopy is discouraged due to the risk of retention at the tumor site, which could lead to small bowel obstruction.
Enterography
CT and MR enterography can help identify small intestinal lesions not visualized by standard imaging techniques. This technique helps assess small bowel NETs beyond the reach of standard endoscopy. MR enterography can also aid in the identification of subtle NET primaries and associated mesenteric fibrosis.[23]
Echocardiography
An echocardiogram should be performed to assess for carcinoid heart disease, particularly in patients with serotonin-secreting tumors, before surgical intervention.[24]
Treatment / Management
Managing GI-NETs depends on the type, location, and tumor stage. Surgery is the mainstay of treatment for patients with localized disease, which can be curative, and those with metastatic disease, which is amenable to cytoreduction. Somatostatin analogs, targeted agents, peptide receptor radioligand therapy, and chemotherapy are used in patients who are not candidates for surgery.
Differential Diagnosis
The differential diagnoses for GI-NETs include:
- Adhesive intestinal obstruction
- Adenocarcinoma
- Lymphoma
- Sarcoma, especially GI stromal tumor
- Nonmalignant polyps (hyperplastic, adenomas)
- Acute appendicitis
Surgical Oncology
Surgical Principles in NET Management
Preoperative Care
All patients undergoing surgery for NETs should be evaluated for the presence of carcinoid syndrome. If carcinoid syndrome is identified, a preoperative transthoracic echocardiogram is essential to assess for carcinoid heart disease, including valvular dysfunction or heart failure. This evaluation is critical to optimizing the patient for surgery and reducing the risk of perioperative complications.[24]
Patients with carcinoid syndrome should receive preoperative treatment with a long-acting somatostatin analog (eg, octreotide or lanreotide) to prevent intraoperative or perioperative carcinoid crisis, characterized by severe flushing, diarrhea, bronchospasm, and hemodynamic instability. For patients undergoing abdominal surgery, particularly when long-term somatostatin analog therapy is planned, a prophylactic cholecystectomy should be considered. Prolonged somatostatin analog use increases the risk of biliary complications such as cholelithiasis or cholecystitis. Prophylactic cholecystectomy can mitigate these risks and prevent the need for subsequent surgical intervention.[25]
Surgical Management of Jejunoileal NETs
Complete surgical resection of the primary tumor with regional lymphadenectomy is the mainstay of managing jejunoileal NET. These tumors are often multifocal, necessitating meticulous intraoperative inspection of the small intestine via manual palpation. Many small lesions are detectable only through this technique, with the majority located in the ileum rather than the jejunum.[25]
Up to 70% of patients with jejunoileal NETs present with lymph node involvement at diagnosis, necessitating a regional lymphadenectomy. Mesenteric resection should be performed when significant mesenteric involvement is present. Cases requiring extensive mesenteric resection have a risk of short bowel syndrome. For these patients, conservative resection with preservation of bowel length may be considered, supplemented with adjunctive therapies. Preservation of the ileocecal valve, when feasible, is advised to minimize the risk of malabsorption and diarrhea. The characteristic desmoplastic reaction associated with NETs often results in mesenteric fibrosis, complicating dissection and increasing the technical challenges of surgery.[25]
For patients with metastatic disease confined to the liver, hepatic resection is indicated to improve symptom control, quality of life, and overall survival. Liver resection should aim to achieve at least 70% to 90% cytoreduction of the tumor burden. The feasibility of liver resection depends on the patient’s performance status, the presence of an adequate functional liver remnant, and the ability to achieve cytoreduction goals. Whenever possible, parenchyma-sparing techniques should be employed to maximize liver function postoperatively.[4][26] For patients with inoperable hepatic metastases, liver-directed therapies can reduce morbidity and improve quality of life. Options include percutaneous hepatic transarterial embolization, radioembolization with yttrium-90 microspheres, and ablation, which are discussed separately.[25]
Surgical Management of Appendiceal NETs
Appendiceal NETs are often diagnosed incidentally during an appendectomy performed for presumed appendicitis. Larger tumors may be identified preoperatively through imaging or other diagnostic evaluations. The surgical management of appendiceal NETs depends on several factors, including the tumor size, depth of invasion, and status of the tumor margins. The rationale for a right hemicolectomy in more advanced appendiceal NET is to perform an adequate lymphadenectomy. A right hemicolectomy is usually not beneficial for patients with low-risk tumors (eg, smaller size, well-differentiated, no transmural invasion).[25] The following management approaches are recommended based on tumor characteristics:
- Tumors sized <1 cm
- No further treatment is required for completely resected tumors with negative margins. However, resection with a possible partial cecectomy is recommended if the margins are positive.
- Tumors sized 1 to 2 cm
- An appendectomy with negative margins is typically sufficient. However, if the tumor exhibits high-risk features, eg, a high Ki-67 index, invasion of the mesoappendix, or a higher histological grade, a completion right hemicolectomy is indicated.
- Tumors >2 cm in size
- A right hemicolectomy is indicated to ensure complete resection and adequate lymphadenectomy.
- Metastatic disease
- The indications for surgery in metastatic disease to the liver are the same as in jejunoileal NET.[4]
Surgical Management of Colonic NETs
Colonic NETs are most commonly identified incidentally during polypectomy specimens, although larger lesions may occasionally present symptomatically.[25] The following management approaches are recommended based on tumor characteristics:
- Lesions <2 cm
- For tumors confined to the mucosa or submucosa without evidence of lymphadenopathy on imaging, endoscopic resection is the treatment of choice if technically feasible.
- Lesions >2 cm or unresectable endoscopically
- Segmental colectomy with lymph node dissection is recommended. The procedure should follow oncologic principles similar to those for colorectal adenocarcinoma to ensure complete resection and proper staging.
When endoscopic resection is performed, the site should be marked (eg, with a tattoo) to facilitate close endoscopic surveillance. Unlike NETs of the small intestine and appendix, cytoreductive surgery or liver-directed interventions are generally not indicated for colonic NETs unless the tumor is hormonally active or causing significant symptoms.
Surgical Management of Rectal NETs
Rectal NETs are frequently identified incidentally during polypectomy or endoscopic evaluations. These lesions are often small and asymptomatic at the time of diagnosis.[25] The following management approaches are recommended based on tumor characteristics:
- Lesions <1 cm in size
- Endoscopic resection is typically adequate for well-differentiated tumors confined to the mucosa or submucosa without evidence of lymphovascular invasion or lymphadenopathy.
- Lesions 1 to 2 cm in size
- Lesions should be evaluated for high-risk features, such as a high Ki-67 index, invasion into deeper layers, or positive margins after resection. If any high-risk features are present, transanal excision or proctectomy may be required.
- Lesions >2 cm or with high-risk features
- Proctectomy with total mesorectal excision is recommended, following the same oncologic principles as rectal adenocarcinoma.
Close endoscopic follow-up is necessary for all patients undergoing resection of rectal NETs to monitor for recurrence or progression. Hormonal or cytoreductive interventions are generally not indicated unless the tumor is hormonally active or causing significant symptoms.
Radiation Oncology
Radiation therapy has a supportive role in the management of GI-NETs. Although these tumors have traditionally been considered relatively radioresistant, radiation therapy can be beneficial in certain clinical scenarios, particularly for palliation of symptoms and local control of metastases.[25] Conventional external beam radiation therapy (EBRT) is most commonly employed for symptomatic relief of localized metastatic lesions or for controlling tumor growth in sites where surgery is not feasible. For instance, EBRT can significantly reduce pain and stabilize skeletal lesions in bone metastases. This therapy may also be considered for palliation of bulky disease in the liver or retroperitoneum, where tumor progression threatens organ function or causes significant morbidity (eg, obstruction, bleeding, or intractable pain).[25][27]
Stereotactic body radiation therapy (SBRT) has emerged as a precise technique for delivering high-dose radiation to well-defined targets over fewer treatment sessions. This approach can be particularly effective for liver or lung metastases from well-differentiated NETs, achieving local control rates comparable to aggressive surgical interventions in select patients. While prospective data on SBRT in NETs remain limited, retrospective series have reported encouraging local control rates and durability of response, often with minimal toxicity.[25]
Selective internal radiation therapy (SIRT), or radioembolization, represents another modality for locoregional management of liver-dominant metastatic NETs. By infusing yttrium-90 microspheres into the hepatic artery, SIRT delivers targeted radiation to liver metastases while minimizing exposure to surrounding healthy tissue. Although randomized data are scarce, retrospective studies have observed meaningful tumor shrinkage, delayed disease progression, and symptomatic improvement in select patients with extensive liver involvement.[25][28]
Medical Oncology
Medical management is indicated in nonresectable, advanced, recurrent, or metastatic NEC. Key therapeutic modalities include somatostatin analogs, mechanistic target of rapamycin (mTOR) inhibitors, peptide receptor radionuclide therapy, and chemotherapy.
Somatostatin Analogs
Somatostatin analogs (eg, octreotide long-acting release [LAR] and lanreotide) are used for symptomatic control in functioning NETs and tumor growth inhibition in well-differentiated (ie, grade 1 or grade 2) tumors. They exert their effects by binding to somatostatin receptors on tumor cells, reducing hormone secretion and alleviating hormone-related symptoms, eg, flushing and diarrhea. Pivotal trials have documented their antiproliferative benefits in delaying tumor progression, most notably the PROMID study in patients with midgut NETs and the CLARINET study in a broader cohort of gastroenteropancreatic NETs. Adverse effects include GI disturbances (eg, steatorrhea and abdominal bloating), biliary lithiasis resulting from reduced gallbladder contractility, and occasional dysglycemia.[25][29][30]
Targeted Therapies with mTOR Inhibitors
Everolimus, an mTOR inhibitor, has shown efficacy in progressive, well-differentiated pancreatic, GI, or pulmonary NETs by blocking a key intracellular pathway responsible for cell growth and angiogenesis. The RADIANT-3 trial established its role in improving progression-free survival in patients with pancreatic NETs. The RADIANT-4 trial demonstrated similar benefits in nonfunctional lung or GI-NETs. Common adverse events include stomatitis, hyperglycemia, dyslipidemia, fatigue, and immunosuppression—all of which necessitate vigilant metabolic and hematologic monitoring.[25][31]
Peptide Receptor Radionuclide Therapy
PRRT employs radiolabeled somatostatin analogs, eg, lutetium-177 (177Lu)-dotatate, to deliver targeted radiation to somatostatin receptor-expressing tumor cells. Please see StatPearls' companion resource, "Neuroendocrine Tumor Lu177Dotatate Therapy", for further information on 177Lu-dotatate therapy. This strategy is effective in advanced, well-differentiated NETs that exhibit high somatostatin receptor density. The NETTER-1 trial significantly improved progression-free survival and quality of life with 177Lu-dotatate compared to high-dose octreotide LAR in metastatic midgut NETs. Although generally well-tolerated, possible toxicities include mild to moderate bone marrow suppression, nephrotoxicity (requiring specific amino acid infusions for renal protection), and transient GI adverse events. Ongoing trials are looking to expand the indications for PRRT.[25][25][32]
Chemotherapy
Chemotherapy is integral in the management of poorly differentiated NECs (World Health Organization grade 3) and certain high-grade NETs, which often display aggressive clinical behavior. Platinum-based regimens, such as cisplatin or carboplatin combined with etoposide, are the standard first-line therapy. Chemotherapy is less frequently used in well-differentiated NETs, although temozolomide-based regimens (sometimes combined with capecitabine) may be beneficial in select cases. Platinum agents can cause nephrotoxicity and ototoxicity, while temozolomide is associated with myelosuppression and GI disturbances.[25]
Prognosis
GI-NETs exhibit a highly variable prognosis, influenced by tumor grade, Ki-67 index, and mitotic count, stage at diagnosis, and histological differentiation. In general, well-differentiated, low- to intermediate-grade NETs tend to have a more indolent course, with 5-year survival rates exceeding 80% for small, localized lesions amenable to complete surgical resection. By contrast, poorly differentiated NECs, especially those presenting with advanced disease, carry a significantly worse outlook, with 5-year survival often falling below 20%. Functional status can also play a role in prognosis, as hormone-producing tumors may be detected earlier due to symptomatic hypersecretion syndromes. Still, aggressive biology can override any potential survival advantage.
Complications
GI-NETs can give rise to various complications affecting quality of life and clinical outcomes. Hormone-related complications are seen with functional GI-NETs, causing carcinoid syndrome and, rarely, carcinoid crisis. Advanced or bulky GI-NETs can cause mechanical complications. Small bowel lesions, for instance, may lead to mesenteric fibrosis, obstruction, and ischemia, whereas colonic or rectal NETs can present with bleeding, altered bowel habits, or significant local mass effects. Metastatic spread, most commonly to the liver, may further compromise hepatic function and exacerbate systemic symptoms through increased hormone release directly into the systemic circulation. In addition, more extensive metastatic deposits can cause pain, organ dysfunction, or secondary complications such as biliary obstruction.
Deterrence and Patient Education
GI-NETs often go unnoticed for extended periods. Therefore, patients must undergo routine check-ups and remain attentive to unusual symptoms such as unexplained flushing, persistent diarrhea, or abdominal pain. Smoking cessation is another valuable step, as it can reduce the chance of many malignancies.
While there is no definitive way to prevent NETs, patients with a relevant family history should communicate concerns to their clinicians, allowing for earlier detection and intervention. Regular follow-up appointments, imaging studies, and recommended blood tests also help identify NETs at an early stage. By staying informed, following healthy lifestyle practices, and promptly reporting new symptoms, patients can help deter complications and optimize the effectiveness of available treatments.
Enhancing Healthcare Team Outcomes
Providing patient-centered care for individuals with gastrointestinal neuroendocrine tumors requires seamless collaboration among diverse healthcare professionals, including medical oncologists, gastroenterologists, endocrinologists, surgeons, advanced practice clinicians, nurses, pharmacists, nuclear medicine specialists, pathologists, and supportive care teams. Clinicians must possess the expertise to accurately diagnose, stage, and manage NETs, using advanced imaging techniques and addressing complications such as carcinoid syndrome or bowel obstruction.
A strategic, evidence-based approach ensures that treatment plans are personalized, balancing the risks and benefits of available therapies. Ethical considerations are integral to this process, emphasizing shared decision-making and respect for patient autonomy. Clearly defined roles and responsibilities within the interprofessional team allow each member to contribute their specialized knowledge—surgical, medical, nutritional, or psychosocial—ensuring a comprehensive approach to optimizing patient outcomes.
Effective interprofessional communication fosters a collaborative environment where timely information exchange occurs and emerging concerns are promptly addressed. Open dialogue between clinicians enhances the continuity of care, reducing medical errors and delays in diagnosis or treatment. Care coordination further streamlines the patient’s journey, integrating various management aspects, from diagnosis to long-term follow-up. This approach ensures patients receive well-organized and efficient care, improving overall safety and satisfaction. By prioritizing integrated teamwork and tailoring care to individual patient needs, healthcare professionals can enhance clinical outcomes, minimize complications, and optimize the overall effectiveness of the care team in managing GI-NETs.
Media
(Click Image to Enlarge)
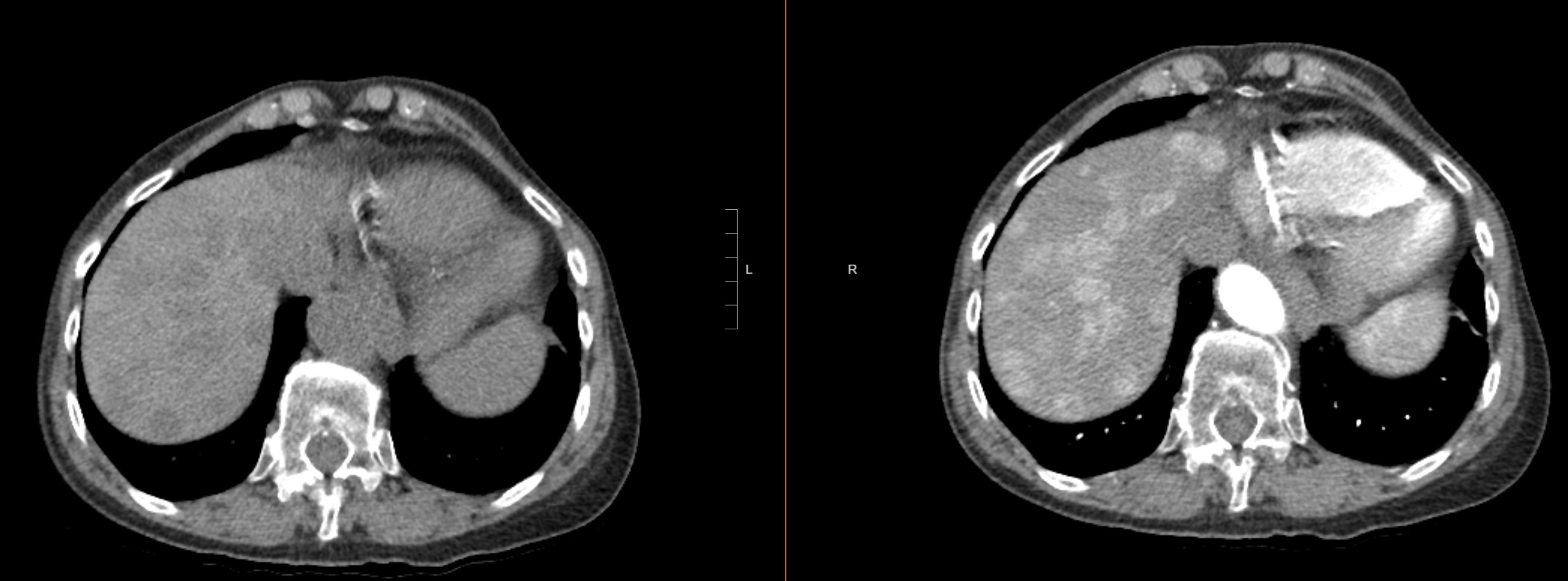
Neuroendocrine Tumor Liver Metastases. Triple-phase computed tomography scan of neuroendocrine tumor liver metastases showing marked arterial enhancement on the arterial phase.
Contributed by G Menon, MD
References
Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A, Evans DB. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008 Jun 20:26(18):3063-72. doi: 10.1200/JCO.2007.15.4377. Epub [PubMed PMID: 18565894]
Level 3 (low-level) evidenceNiederle B, Pape UF, Costa F, Gross D, Kelestimur F, Knigge U, Öberg K, Pavel M, Perren A, Toumpanakis C, O'Connor J, O'Toole D, Krenning E, Reed N, Kianmanesh R, Vienna Consensus Conference participants. ENETS Consensus Guidelines Update for Neuroendocrine Neoplasms of the Jejunum and Ileum. Neuroendocrinology. 2016:103(2):125-38. doi: 10.1159/000443170. Epub 2016 Jan 12 [PubMed PMID: 26758972]
Level 3 (low-level) evidenceRamesh A, Chatterjee A, Subramaniam RM. Neuroendocrine Neoplasms: Epidemiology, Diagnosis, and Management. PET clinics. 2023 Apr:18(2):161-168. doi: 10.1016/j.cpet.2022.10.002. Epub 2023 Jan 25 [PubMed PMID: 36707369]
Pavel M, Öberg K, Falconi M, Krenning EP, Sundin A, Perren A, Berruti A, ESMO Guidelines Committee. Electronic address: clinicalguidelines@esmo.org. Gastroenteropancreatic neuroendocrine neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology : official journal of the European Society for Medical Oncology. 2020 Jul:31(7):844-860. doi: 10.1016/j.annonc.2020.03.304. Epub 2020 Apr 6 [PubMed PMID: 32272208]
Level 1 (high-level) evidenceDas S, Dasari A. Epidemiology, Incidence, and Prevalence of Neuroendocrine Neoplasms: Are There Global Differences? Current oncology reports. 2021 Mar 14:23(4):43. doi: 10.1007/s11912-021-01029-7. Epub 2021 Mar 14 [PubMed PMID: 33719003]
Rindi G, Mete O, Uccella S, Basturk O, La Rosa S, Brosens LAA, Ezzat S, de Herder WW, Klimstra DS, Papotti M, Asa SL. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocrine pathology. 2022 Mar:33(1):115-154. doi: 10.1007/s12022-022-09708-2. Epub 2022 Mar 16 [PubMed PMID: 35294740]
Level 3 (low-level) evidenceOronsky B, Ma PC, Morgensztern D, Carter CA. Nothing But NET: A Review of Neuroendocrine Tumors and Carcinomas. Neoplasia (New York, N.Y.). 2017 Dec:19(12):991-1002. doi: 10.1016/j.neo.2017.09.002. Epub 2017 Nov 5 [PubMed PMID: 29091800]
Clift AK, Kidd M, Bodei L, Toumpanakis C, Baum RP, Oberg K, Modlin IM, Frilling A. Neuroendocrine Neoplasms of the Small Bowel and Pancreas. Neuroendocrinology. 2020:110(6):444-476. doi: 10.1159/000503721. Epub 2019 Sep 27 [PubMed PMID: 31557758]
Feingold KR, Ahmed SF, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, Levy M, McGee EA, McLachlan R, Muzumdar R, Purnell J, Rey R, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, Hofland J, de Herder WW. Gastrointestinal Neuroendocrine Tumors and the Carcinoid Syndrome. Endotext. 2000:(): [PubMed PMID: 25905385]
Kidd M, Bodei L, Modlin IM. Chromogranin A: any relevance in neuroendocrine tumors? Current opinion in endocrinology, diabetes, and obesity. 2016 Feb:23(1):28-37. doi: 10.1097/MED.0000000000000215. Epub [PubMed PMID: 26627724]
Level 3 (low-level) evidenceNguyen M, Li M, Travers A, Segelov E. Role of Chromogranin A in the Diagnosis and Follow-up of Neuroendocrine Tumors: Real-World Experience. Pancreas. 2022 Sep 1:51(8):1007-1010. doi: 10.1097/MPA.0000000000002132. Epub [PubMed PMID: 36607947]
Baekdal J, Krogh J, Klose M, Holmager P, Langer SW, Oturai P, Kjaer A, Federspiel B, Hilsted L, Rehfeld JF, Knigge U, Andreassen M. Limited Diagnostic Utility of Chromogranin A Measurements in Workup of Neuroendocrine Tumors. Diagnostics (Basel, Switzerland). 2020 Oct 29:10(11):. doi: 10.3390/diagnostics10110881. Epub 2020 Oct 29 [PubMed PMID: 33138020]
de Mestier L, Savagner F, Brixi H, Do Cao C, Dominguez-Tinajero S, Roquin G, Goichot B, Hentic O, Dubreuil O, Hautefeuille V, Walter T, Cadiot G. Plasmatic and Urinary 5-Hydroxyindolacetic Acid Measurements in Patients With Midgut Neuroendocrine Tumors: A GTE Study. The Journal of clinical endocrinology and metabolism. 2021 Mar 25:106(4):e1673-e1682. doi: 10.1210/clinem/dgaa924. Epub [PubMed PMID: 33382891]
Malczewska A, Kos-Kudła B, Kidd M, Drozdov I, Bodei L, Matar S, Oberg K, Modlin IM. The clinical applications of a multigene liquid biopsy (NETest) in neuroendocrine tumors. Advances in medical sciences. 2020 Mar:65(1):18-29. doi: 10.1016/j.advms.2019.10.002. Epub 2019 Dec 13 [PubMed PMID: 31841822]
Level 3 (low-level) evidenceModlin IM, Kidd M, Frilling A, Falconi M, Filosso PL, Malczewska A, Kitz A. Molecular Genomic Assessment Using a Blood-based mRNA Signature (NETest) is Cost-effective and Predicts Neuroendocrine Tumor Recurrence With 94% Accuracy. Annals of surgery. 2021 Sep 1:274(3):481-490. doi: 10.1097/SLA.0000000000005026. Epub [PubMed PMID: 34183517]
Komarnicki P, Musiałkiewicz J, Stańska A, Maciejewski A, Gut P, Mastorakos G, Ruchała M. Circulating Neuroendocrine Tumor Biomarkers: Past, Present and Future. Journal of clinical medicine. 2022 Sep 21:11(19):. doi: 10.3390/jcm11195542. Epub 2022 Sep 21 [PubMed PMID: 36233409]
Dromain C, de Baere T, Lumbroso J, Caillet H, Laplanche A, Boige V, Ducreux M, Duvillard P, Elias D, Schlumberger M, Sigal R, Baudin E. Detection of liver metastases from endocrine tumors: a prospective comparison of somatostatin receptor scintigraphy, computed tomography, and magnetic resonance imaging. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005 Jan 1:23(1):70-8 [PubMed PMID: 15625361]
Level 1 (high-level) evidenceMaxwell JE, Howe JR. Imaging in neuroendocrine tumors: an update for the clinician. International journal of endocrine oncology. 2015:2(2):159-168 [PubMed PMID: 26257863]
Bader TR, Semelka RC, Chiu VC, Armao DM, Woosley JT. MRI of carcinoid tumors: spectrum of appearances in the gastrointestinal tract and liver. Journal of magnetic resonance imaging : JMRI. 2001 Sep:14(3):261-9 [PubMed PMID: 11536403]
Kapoor M, Kasi A. Octreotide Scan. StatPearls. 2025 Jan:(): [PubMed PMID: 32644756]
Galgano SJ, Sharbidre K, Morgan DE. Multimodality Imaging of Neuroendocrine Tumors. Radiologic clinics of North America. 2020 Nov:58(6):1147-1159. doi: 10.1016/j.rcl.2020.07.008. Epub 2020 Sep 11 [PubMed PMID: 33040854]
Sakellis C, Jacene HA. Neuroendocrine Tumors: Diagnostics. PET clinics. 2024 Jul:19(3):325-339. doi: 10.1016/j.cpet.2024.03.008. Epub 2024 May 6 [PubMed PMID: 38714399]
Vlachou E, Koffas A, Toumpanakis C, Keuchel M. Updates in the diagnosis and management of small-bowel tumors. Best practice & research. Clinical gastroenterology. 2023 Jun-Aug:64-65():101860. doi: 10.1016/j.bpg.2023.101860. Epub 2023 Aug 12 [PubMed PMID: 37652650]
Jin C, Sharma AN, Thevakumar B, Majid M, Al Chalaby S, Takahashi N, Tanious A, Arockiam AD, Beri N, Amsterdam EA. Carcinoid Heart Disease: Pathophysiology, Pathology, Clinical Manifestations, and Management. Cardiology. 2021:146(1):65-73. doi: 10.1159/000507847. Epub 2020 Oct 16 [PubMed PMID: 33070143]
Shah MH, Goldner WS, Benson AB, Bergsland E, Blaszkowsky LS, Brock P, Chan J, Das S, Dickson PV, Fanta P, Giordano T, Halfdanarson TR, Halperin D, He J, Heaney A, Heslin MJ, Kandeel F, Kardan A, Khan SA, Kuvshinoff BW, Lieu C, Miller K, Pillarisetty VG, Reidy D, Salgado SA, Shaheen S, Soares HP, Soulen MC, Strosberg JR, Sussman CR, Trikalinos NA, Uboha NA, Vijayvergia N, Wong T, Lynn B, Hochstetler C. Neuroendocrine and Adrenal Tumors, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network : JNCCN. 2021 Jul 28:19(7):839-868. doi: 10.6004/jnccn.2021.0032. Epub 2021 Jul 28 [PubMed PMID: 34340212]
Level 1 (high-level) evidenceFrilling A, Modlin IM, Kidd M, Russell C, Breitenstein S, Salem R, Kwekkeboom D, Lau WY, Klersy C, Vilgrain V, Davidson B, Siegler M, Caplin M, Solcia E, Schilsky R, Working Group on Neuroendocrine Liver Metastases. Recommendations for management of patients with neuroendocrine liver metastases. The Lancet. Oncology. 2014 Jan:15(1):e8-21. doi: 10.1016/S1470-2045(13)70362-0. Epub [PubMed PMID: 24384494]
Loree JM. Radiating the "Radio-Resistant": The Misunderstood Story of Neuroendocrine Tumors and Radiation. International journal of radiation oncology, biology, physics. 2023 Nov 15:117(4):785. doi: 10.1016/j.ijrobp.2023.01.039. Epub [PubMed PMID: 37838447]
Lewandowski RJ, Toskich BB, Brown DB, El-Haddad G, Padia SA. Role of Radioembolization in Metastatic Neuroendocrine Tumors. Cardiovascular and interventional radiology. 2022 Nov:45(11):1590-1598. doi: 10.1007/s00270-022-03206-y. Epub 2022 Aug 2 [PubMed PMID: 35918431]
Caplin ME, Pavel M, Phan AT, Ćwikła JB, Sedláčková E, Thanh XT, Wolin EM, Ruszniewski P, CLARINET Investigators. Lanreotide autogel/depot in advanced enteropancreatic neuroendocrine tumours: final results of the CLARINET open-label extension study. Endocrine. 2021 Feb:71(2):502-513. doi: 10.1007/s12020-020-02475-2. Epub 2020 Oct 14 [PubMed PMID: 33052555]
Rinke A, Müller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, Mayer C, Aminossadati B, Pape UF, Bläker M, Harder J, Arnold C, Gress T, Arnold R, PROMID Study Group. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009 Oct 1:27(28):4656-63. doi: 10.1200/JCO.2009.22.8510. Epub 2009 Aug 24 [PubMed PMID: 19704057]
Level 1 (high-level) evidenceYao JC, Fazio N, Singh S, Buzzoni R, Carnaghi C, Wolin E, Tomasek J, Raderer M, Lahner H, Voi M, Pacaud LB, Rouyrre N, Sachs C, Valle JW, Fave GD, Van Cutsem E, Tesselaar M, Shimada Y, Oh DY, Strosberg J, Kulke MH, Pavel ME, RAD001 in Advanced Neuroendocrine Tumours, Fourth Trial (RADIANT-4) Study Group. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet (London, England). 2016 Mar 5:387(10022):968-977. doi: 10.1016/S0140-6736(15)00817-X. Epub 2015 Dec 17 [PubMed PMID: 26703889]
Level 1 (high-level) evidenceStrosberg JR, Caplin ME, Kunz PL, Ruszniewski PB, Bodei L, Hendifar A, Mittra E, Wolin EM, Yao JC, Pavel ME, Grande E, Van Cutsem E, Seregni E, Duarte H, Gericke G, Bartalotta A, Mariani MF, Demange A, Mutevelic S, Krenning EP, NETTER-1 investigators. (177)Lu-Dotatate plus long-acting octreotide versus high‑dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. The Lancet. Oncology. 2021 Dec:22(12):1752-1763. doi: 10.1016/S1470-2045(21)00572-6. Epub 2021 Nov 15 [PubMed PMID: 34793718]
Level 1 (high-level) evidence