Introduction
Craniosynostosis occurs due to the premature in-utero fusion of one or more cranial sutures. These sutures allow for passage through the birth canal and, later in development, allow the expansion and growth of the underlying brain. When these sutures close prematurely, the head shape becomes altered depending on the sutures involved. Most cases involve a single suture and are classified as non-syndromic. However, Crouzon, Pfeiffer, and Apert syndromes have been associated with multi-sutural craniosynostosis.[1] The treatment of craniosynostosis involves surgical intervention to unlock the fused sutures, ensuring unrestricted brain development and correcting cosmetic deformities.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Almost 20% of all craniosynostoses have a genetic basis, most of which are inherited in an autosomal dominant fashion.[2] Roughly 86% of cases involve a single-gene mutation, whereas 15% are linked to chromosomal abnormalities.[3][4] The most common genes affected in craniosynostosis are those in the fibroblast growth factor receptor (FGFR) pathway. FGFR-1 is associated with Pfeiffer syndrome, FGFR-2 with Apert, Crouzon, and type 1 Pfeiffer syndromes, and FGFR-3 with Muenke syndrome.[3][5] Mutations in TWIST1 have been described in cases of Saethre-Chotzen syndrome.[3][6] Although most cases with these mutations are found in syndromic cases, mutations in SMAD6, TCF12, ERF, and MSX2 have been associated with non-syndromic cases.[7]
Epidemiology
The prevalence of craniosynostosis is 1 in 2000 to 2500 live births.[8][7] Risk factors include maternal smoking, in utero exposure to teratogens, intrauterine constraint, diabetes, and excessive use of caffeine. Thyroid disease may also predispose to developing craniosynostosis.[9] A well-known risk factor for developing craniosynostosis is the presence of a ventriculoperitoneal shunt.[10][11] A study found that nearly half of patients developed craniosynostosis after shunting, with the sagittal suture being most commonly affected.[12]
Non-syndromic craniosynostosis occurs in roughly 75% of cases, with the remaining cases observed with syndromic craniosynostosis.[13] Craniosynostosis is classified based on the affected suture—sagittal craniosynostosis occurs in 55% to 60% of the cases, unilateral coronal craniosynostosis in 20% to 25% of cases, metopic craniosynostosis in approximately 15% of cases, and lambdoid craniosynostosis in 3% to 5% of cases. Clinical identification is typically apparent within the first year of life.[3][13]
Pathophysiology
Some of the risk factors identified to contribute to the development of craniosynostosis include:
- Environmental factors: Advanced parental age, maternal smoking (≥15 cigarettes/day), in vitro fertilization, and specific medications, such as valproic acid, nitrofurantoin, and sertraline.
- Nutritional factors: Certain nutrients, such as vitamins B6, E, and C, lower the risk of specific sutural synostosis, whereas others, such as choline and vitamin B12, may increase the risk of metopic craniosynostosis.
- Thyroid dysfunction: Maternal thyroid dysfunction increases the risk of single-suture synostosis, particularly affecting the sagittal suture. Thyroid disease is a modifiable risk factor.
- Metabolic bone disorders: Conditions such as rickets, hypophosphatasia, osteopetrosis, and mucopolysaccharidosis are associated with secondary craniosynostosis. These disorders often present later with variable suture involvement.
- Mechanical forces: Abnormal brain growth; intrauterine constraints, such as multiple births and bicornuate uterus; and conditions, such as hypoxic-ischemic encephalopathy, contribute to single-suture synostosis.
- Genetic factors: Around 15% of SSC cases involve causal variants in 29 genes, with mutations being autosomal dominant in 8% of cases. Gene associations vary by suture type and familial patterns.
- Gender and ethnic variability: Boys are more likely to have sagittal and metopic craniosynostosis, whereas girls are more prone to unilateral coronal synostosis. Ethnic differences in incidence have been observed, with a higher prevalence of metopic craniosynostosis in Caucasians.[14]
History and Physical
Obtaining a detailed history and performing a thorough physical examination are crucial in determining the diagnosis, as the different types of craniosynostosis have patterned head shapes. While obtaining the history, it is important to inquire about a family history of craniosynostosis, in utero exposure to teratogenic drugs, intrauterine restraints, abnormal fetal positioning, complications during pregnancy, and any delayed milestones. The physical examination allows the clinician to evaluate for craniosynostosis and whether any other features suggest the presence of an underlying syndrome.
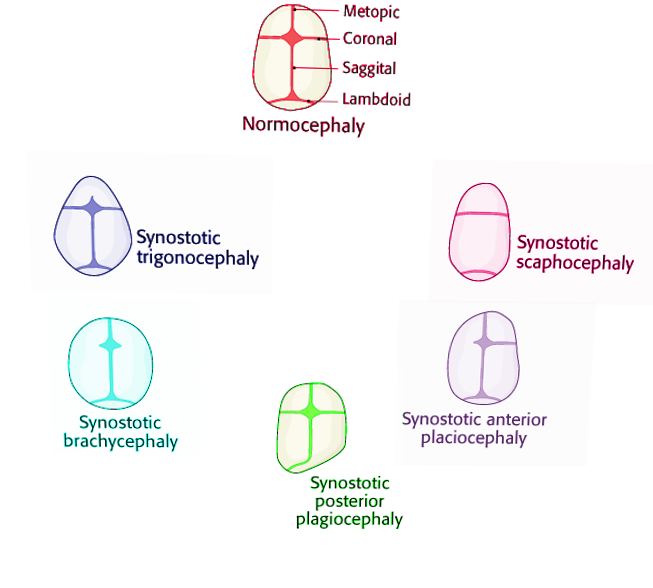
Assessing head shape is the most important diagnostic tool, as the different types of craniosynostosis have classic patterns that can be easily recognized. According to Virchow's law, the cranium grows parallel to the fused suture with growth restriction in the direction perpendicular to the fused suture. This principle accounts for the characteristic head shapes observed in the different types of craniosynostosis as described below:
- Scaphocephaly or dolichocephaly (premature fusion of the sagittal suture): The presentation includes a long, narrow, boat-shaped head with a greater anteroposterior diameter. Frontal and occipital bossing is often present.[15] A palpable ridge may be present along the sagittal suture. The fontanelle may feel more triangular rather than diamond-shaped.
- Clinocephaly (saddle head): Clinocephaly is typically a late presentation of sagittal suture synostosis, characterized by a retro-coronal concavity along the midvault.[16]
- Anterior plagiocephaly (premature fusion of a single coronal suture): The forehead appears flattened on the affected side, with a Harlequin eye deformity characterized by ipsilateral elevation of the sphenoid wing and orbital roof visible on a frontal X-ray. Additional features include frontal bossing on the unaffected side and nasal root deviation toward the side of the fused suture.[15] Compensatory bossing of the contralateral side with chin deviation toward the unaffected side is observed. The ipsilateral eye appears larger, and the eyebrow is raised. The ear is anteriorly displaced on the affected side, distinguishing it from deformational plagiocephaly.
- Posterior plagiocephaly (premature fusion of a single lambdoid suture): This condition presents with ipsilateral flattening of the parietal bone and occiput, a prominent mastoid process, and the ipsilateral ear is posterior and inferior compared to the normal side.[15] A tilted skull base with facial asymmetry is observed as the child grows. There is a significant association with Chiari malformations.[17]
- Pachycephaly (premature fusion of bilateral lambdoid sutures): This condition leads to a total flatness of the back of the skull.[18]
- Trigonocephaly (premature fusion of the metopic suture): The forehead is narrow and pointed; the head has a triangular shape viewed from above, and hypotelorism is often present.[15] Raised or arched eyebrows and lateral orbital hypoplasia are commonly observed.
- Brachycephaly (bi-coronal fusion): This condition has a shortened anteroposterior and widened transverse diameters.[15]
- Oxycephaly (turricephaly, acrocephaly, or high-head syndrome): This condition occurs due to the fusion of more than one suture and results in a towering appearance.
- Kleeblattschadel (cloverleaf skull): A rare deformity caused by the premature fusion of multiple cranial sutures, leading to a trilobar skull shape with frontal bossing, temporal bulging, and a flat posterior skull. The coronal and lambdoid sutures are most commonly affected, though fusion patterns can vary. This condition is typically associated with cosmetic facial deformity, micromyelia (small limb development), increased intracranial pressure, hydrocephalus, hindbrain herniation, skull base dysplasia, and neurological dysfunction.[19]
Other conditions frequently associated with craniosynostosis include hydrocephalus and Chiari I malformations.[20] Hydrocephalus is often observed in cases involving multiple sutures and syndromic craniosynostosis,[20][21][22] with higher rates reported in Crouzon syndrome compared to other craniofacial syndromes.[22] Chiari I malformations are also well-described in the literature and are most frequently associated with lambdoid synostosis,[23][24][17] but they can be observed in other subtypes, such as sagittal synostosis.[25]
The most common syndromes associated with craniosynostosis are described below:
- Apert syndrome: Apert syndrome is characterized by bi-coronal synostosis and turribrachycephaly. This syndrome presents with midface hypoplasia, hypertelorism, down-slanting palpebral fissures, cleft palate, and pseudoprognathic mandible. Other findings include syndactyly of hands and feet, and a short and broad thumb that is often radially deviated. Individuals with Apert syndrome may exhibit mild-to-moderate intellectual disabilities.[15][26][27][28]
- Crouzon syndrome: Crouzon syndrome affects coronal, sagittal, or lambdoid sutures. Key features include midface hypoplasia, a beaked nose, exophthalmos, hypertelorism, and cervical vertebral fusion. Some patients can present with cleft lip or palate. These patients typically have normal intelligence.[15][26][27][28]
- Pfeiffer syndrome: Pfeiffer syndrome features turribrachycephaly in type 1 and Kleeblattschadel in types II and III. Other features include hypertelorism, maxillary hypoplasia, broad thumbs, great toe, syndactyly, and brachydactyly.[15][28]
- Muenke syndrome: Muenke syndrome primarily affects the coronal suture (unilateral or bilateral). Key features include intellectual disability and hearing loss.[15][27][28]
- Kleeblattschadel syndrome (cloverleaf deformity): Kleeblattschadel syndrome is caused by the synostosis of the coronal and lambdoid sutures, resulting in a tri-lobar-shaped head. Characteristic features include a beak-shaped nose, maxillary hypoplasia with proptosis, and inferiorly displaced ears. This type of craniosynostosis is associated with hydrocephalus and is most commonly observed in Pfeiffer syndrome.[29]
Evaluation
Although craniosynostosis is primarily diagnosed clinically, radiologic imaging is often used to support and confirm the diagnosis.
Plain x-rays are low-cost and readily available in most treatment centers. X-rays can help confirm the diagnosis of craniosynostosis when the clinical examination is not definitive. Common findings on x-rays include perisutural sclerosis, bony bridging, and loss of suture clarity. Other findings that can often be used to identify craniosynostosis include the presence of a Harlequin eye (coronal synostosis), deviation of the posterior fontanelle (unilateral lambdoid), or ovoid-shaped and upward-angled orbits (metopic).[30] X-rays have been reported to have a high specificity, but the sensitivity is relatively poor. Cranial ultrasound is another imaging modality that can be used. Ultrasounds are often dependent on the operator but can help determine whether the underlying suture is patent. Patent sutures show a hypoechoic gap, whereas fused sutures do not.
The gold standard for imaging is a computed tomography (CT) scan with three-dimensional reconstructions, which readily assess all sutures. CT scans can also help assess the ventricles in cases with underlying hydrocephalus.
Magnetic resonance imaging is less effective compared to CT in diagnosing craniosynostosis; however, it can aid in evaluating hydrocephalus, Chiari malformations, and other intracranial abnormalities.
If a syndromic case is suspected, genetic testing should be performed, particularly for FGFR gene mutations. Ongoing research continues to explore and identify the genetic causes of these syndromes. To date, 57 genes have been identified as having a relationship with craniosynostosis, the most common ones being those mentioned previously.[13][31][27]
Due to the risk of developing elevated intracranial pressure, especially in cases of syndromic craniosynostosis,[32][33] intracranial pressure monitoring may be indicated. Elevated intracranial pressure results from underlying hydrocephalus, osseous changes of the skull base affecting venous outflow, and midface hypoplasia resulting in sleep apnea. Screening for elevated intracranial pressure begins with an ophthalmologic evaluation to assess for papilledema. In addition to screening for papilledema, optical coherence tomography is effective in identifying elevated intracranial pressure.[34][35] However, if these modalities are inconclusive, intracranial pressure monitoring is indicated.[36]
In patients with suspected sleep-disordered breathing, polysomnography should be recommended to evaluate for sleep apnea.[37][38]
Hearing evaluations should also be performed, as hearing loss can occur in both syndromic and non-syndromic cases [39] due to middle ear effusions. Most often, hearing loss is conductive; however, in cases of Muenke syndrome, hearing loss is more often sensorineural in origin.[40]
Treatment / Management
Immediate Concerns in the Neonatal Period
- Airway management: Support may be necessary for airway obstruction, particularly in cases of multisuture craniosynostosis or syndromic craniosynostosis.
- Emergency measures include continuous positive airway pressure (CPAP), nasal airway devices, or intubation.
- In some cases, a tracheostomy may be required until surgical correction is performed.
- Primary treatment: Craniofacial surgery is the cornerstone of treatment for craniosynostosis.
Indications for Surgical Treatment
- Risk of increased intracranial pressure
- Abnormal skull shape
- Correction of cosmetic deformity
- Expanding intracranial volume to allow for sufficient brain growth
- Protection of the globes in cases of midface retrusion
- Syndromic craniosynostosis often requires a complex, staged surgical approach to address associated deformities and functional abnormalities. This type of surgery may involve significant blood loss and carries a risk of massive hemorrhage.
Types of Procedures
Minimally invasive (endoscopic) surgery:
- Favored for its lower risk of blood transfusion and less noticeable scarring.
- Ideally performed between 3 and 4 months, but no later than 6 months, as effectiveness decreases with age.
- Surgery is not recommended before 3 months due to anesthesia-related risks.
- Requires postoperative helmet therapy worn 23 hours daily for up to 1 year.
- Advantages when compared to open cranial vault repairs:
- Shorter operation times
- Shorter anesthesia times
- Lower transfusion rates
- Decreased length of stay [41]
Open cranial vault reconstruction:
- Typically performed between 6 and 12 months of age or if the patient is referred after 6 months.
- Carries a higher risk of blood loss and transfusion compared to endoscopic surgery.
- The optimal timing for surgery is before the child reaches 1 year, depending on factors such as:
- Child's age at presentation
- The presence of functional issues such as increased intracranial pressure or airway obstruction
- Type and severity of craniosynostosis
- Associated comorbidities
- Preferences of the surgical team
- Specific recommendation: Surgery for sagittal suture synostosis is best performed before 6 months to achieve optimal outcomes.
The management of craniosynostosis primarily focuses on surgical intervention. For cases involving a single suture, surgery is often performed in an elective manner. However, in cases of multi-sutural involvement, surgery may be expedited if there is concern for elevated intracranial pressure. In cases of sagittal synostosis, spring-mediated cranioplasty may be used in addition to suturectomy.[42][43] A limitation of using springs is the necessity for a second surgery to remove them once the desired head shape is accomplished. Once the child reaches the age of 6 months, endoscopic suturectomy is typically not performed because the skull becomes too hard for postoperative helmeting to be effective. In these cases, more extensive surgeries are required, including cranial vault reconstructions, frontal-orbital advancements, or distraction osteogenesis, depending on the subtype of craniosynostosis.
If patients develop hydrocephalus, treatment options involve either placement of a shunt or endoscopic third ventriculostomy, with or without choroid plexus coagulation.[22] When considering a ventriculoperitoneal shunt, it is essential to consider the need for further open vault reconstructions. Placement of a frontal shunt may result in exposure of the shunt hardware, making a posterior approach potentially more appropriate. If there is a concern for the need for future operations, an endoscopic third ventriculostomy may be a reasonable initial approach.
Differential Diagnosis
The primary differential diagnosis for craniosynostosis includes deformational or positional plagiocephaly, which can appear similar to lambdoid craniosynostosis with flattening of the occiput. Positional plagiocephaly presents with a parallelogram-shaped head, anterior displacement of the ipsilateral ear, and ipsilateral occipital flattening with contralateral occipital bossing.[26][27] The prevalence has increased over time due to the Back-to-Sleep campaign to reduce the incidence of sudden infant death syndrome. However, the only concern is cosmetic, and it can be managed supportively by alternating the side of the head on which the baby sleeps, and if refractory, remodeling helmets are an option. This condition does not require surgical intervention and does not affect neurologic development.[13][44][3]
In cases of metopic craniosynostosis, the differential diagnosis includes benign metopic ridging, which has been described as an intermediate phenotype between metopic craniosynostosis and normal anatomy [45][46] and does not require surgical intervention. True metopic craniosynostosis typically has a narrow forehead, biparietal widening, and hypotelorism. A retrospective study found a narrow forehead and pterional constriction in all patients with metopic craniosynostosis but in only 11.2% and 2.8% of patients with metopic ridging, respectively. The study also found that patients with metopic craniosynostosis were more likely to present before 6 months compared to those with benign metopic ridging.[47]
Torticollis is another condition in which patients may develop cranial and facial asymmetry similar to those observed with craniosynostosis. Most often, patients with torticollis sleep on their backs with their heads on the same side, which can precipitate deformational plagiocephaly and appear similar to lambdoid craniosynostosis.[48][49]
Treatment Planning
Preoperative planning typically includes a three-dimensional CT scan, which can be used to create templates for the accurate placement of osteotomies.[50] [51][52]
Prognosis
If left untreated, craniosynostosis can affect development due to the growth restriction of the brain and increased intracranial pressure. Children with single-suture synostosis have less developmental delay compared to children with multi-sutural or syndromic craniosynostosis, with delays reported in up to 35% to 50% of children with single-suture craniosynostosis.[53] Although based on a small sample size, a study examining the neurodevelopmental outcomes in patients with metopic craniosynostosis found that, overall, these individuals had an above-average intelligence quotient and academic achievement close to the national average. The study also found a correlation between the preoperative radiographic severity and the decline in performance.[54] Another study showed children with metopic craniosynostosis have more difficulties with executive function compared to those with other subtypes of single-suture craniosynostosis.[55] Further studies have demonstrated subtle differences in achievement, although patients fall within the normal range of IQ.[56] Other studies have shown consistently lower neurodevelopmental scores in patients with single-suture craniosynostosis compared to those without craniosynostosis.[57]
Follow-up is recommended to determine the need for further surgical correction. A study found that up to 8.8% of patients were found to have secondary craniosynostosis of sutures that were initially patent before the first operation.[58] Furthermore, patients should be followed by ophthalmology to screen for papilledema. With early identification of developmental gaps and placement in support programs, a negative academic and cognitive outcome may be reduced.[13][59]
Complications
As with any surgical intervention, these cases carry inherent risks. A retrospective cohort study reported complication rates in 295 patients who underwent both endoscopic and open procedures. In non-syndromic cases, surgical complications were reported in 1.3% of cases, whereas medical complications were reported in 4.5% of cases. Intra-operative durotomies were reported in 3.6% of the endoscopic cases and 7.8% of the open cases. Rates of complications were similar between the non-syndromic and syndromic cases.[60]
Consultations
When patients are diagnosed with craniosynostosis, the following consultations should be considered:
- Pediatric neurosurgeons
- Plastic surgeons and facial plastic surgeons, ideally those who have had specialized training in pediatric craniofacial abnormalities
- Oral and maxillofacial surgeons
- Developmental pediatricians
- Genetic specialists
- Pediatric otolaryngologists
- Sleep medicine specialists
- Pediatric ophthalmologists
- Speech-language pathologists
- Pediatric anesthesiologists
- Audiologists
- Orthodontists and pediatric dentists
- Psychologists and psychiatrists
- Social workers
- Feeding specialists
Deterrence and Patient Education
Parents should be educated about the causes of positional plagiocephaly and strategies to reduce the risk of developing plagiocephaly, such as alternating the side on which the baby sleeps.
Pearls and Other Issues
Craniosynostosis is a condition characterized by the premature fusion of one or more cranial sutures, and its management requires a comprehensive understanding of its types, causes, and treatment approaches, which are as follows:
- Craniosynostosis results from the premature fusion of one or more cranial sutures.
- This condition is commonly observed in 1 per 2000 to 2500 children.
- Craniosynostosis is categorized into syndromic and non-syndromic types.
- The most common non-syndromic subtype is scaphocephaly.
- In syndromic cases, the most commonly affected gene is the FGFR gene.
- Surgical intervention is the preferred treatment. The surgical approach typically depends on the child's age and involves sutures.
- Positional plagiocephaly does not require surgical therapy.
- An interprofessional approach to care is essential in treating patients.
Enhancing Healthcare Team Outcomes
An interprofessional approach to care is essential in treating patients with craniosynostosis, especially those with syndromic craniosynostosis. A coordinated team of healthcare professionals ensures comprehensive treatment and addresses the multiple aspects of the condition. The team should include pediatricians, neurosurgeons, plastic surgeons, otolaryngologists, maxillofacial surgeons, ophthalmologists, geneticists, nurses, sleep medicine specialists, respiratory physicians, and developmental pediatricians. Each specialist plays a vital role in diagnosing, treating, and monitoring the patient's progress, addressing both the cranial and systemic aspects of the condition.
Collaboration among these professionals allows for individualized care plans that consider the child's age, the severity of the condition, and any associated syndromes. Early involvement of genetic counseling can help assess the risk of recurrence and guide family planning, whereas ophthalmologists and sleep specialists monitor potential complications related to vision and breathing. Neurosurgeons and plastic surgeons focus on correcting cranial deformities, whereas developmental specialists provide ongoing support for cognitive and motor skills. This holistic, multidisciplinary approach not only improves surgical outcomes but also enhances long-term developmental and quality-of-life prospects for patients with craniosynostosis.
Media
(Click Image to Enlarge)
Craniosynostosis Image courtesy Dr Chaigasame
References
Hersh DS, Hughes CD. Syndromic Craniosynostosis: Unique Management Considerations. Neurosurgery clinics of North America. 2022 Jan:33(1):105-112. doi: 10.1016/j.nec.2021.09.008. Epub 2021 Oct 26 [PubMed PMID: 34801135]
Lajeunie E, Crimmins DW, Arnaud E, Renier D. Genetic considerations in nonsyndromic midline craniosynostoses: a study of twins and their families. Journal of neurosurgery. 2005 Oct:103(4 Suppl):353-6 [PubMed PMID: 16270687]
Johnson D, Wilkie AO. Craniosynostosis. European journal of human genetics : EJHG. 2011 Apr:19(4):369-76. doi: 10.1038/ejhg.2010.235. Epub 2011 Jan 19 [PubMed PMID: 21248745]
Wilkie AO, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, Lester T, Richards PG, Twigg SR, Wall SA. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010 Aug:126(2):e391-400. doi: 10.1542/peds.2009-3491. Epub 2010 Jul 19 [PubMed PMID: 20643727]
Azoury SC, Reddy S, Shukla V, Deng CX. Fibroblast Growth Factor Receptor 2 (FGFR2) Mutation Related Syndromic Craniosynostosis. International journal of biological sciences. 2017:13(12):1479-1488. doi: 10.7150/ijbs.22373. Epub 2017 Nov 2 [PubMed PMID: 29230096]
Kress W, Schropp C, Lieb G, Petersen B, Büsse-Ratzka M, Kunz J, Reinhart E, Schäfer WD, Sold J, Hoppe F, Pahnke J, Trusen A, Sörensen N, Krauss J, Collmann H. Saethre-Chotzen syndrome caused by TWIST 1 gene mutations: functional differentiation from Muenke coronal synostosis syndrome. European journal of human genetics : EJHG. 2006 Jan:14(1):39-48 [PubMed PMID: 16251895]
Timberlake AT, Persing JA. Genetics of Nonsyndromic Craniosynostosis. Plastic and reconstructive surgery. 2018 Jun:141(6):1508-1516. doi: 10.1097/PRS.0000000000004374. Epub [PubMed PMID: 29579021]
Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989-2003. American journal of medical genetics. Part A. 2008 Apr 15:146A(8):984-91. doi: 10.1002/ajmg.a.32208. Epub [PubMed PMID: 18344207]
Stanton E, Urata M, Chen JF, Chai Y. The clinical manifestations, molecular mechanisms and treatment of craniosynostosis. Disease models & mechanisms. 2022 Apr 1:15(4):. doi: 10.1242/dmm.049390. Epub 2022 Apr 22 [PubMed PMID: 35451466]
Faulhauer K, Schmitz P. Overdrainage phenomena in shunt treated hydrocephalus. Acta neurochirurgica. 1978:45(1-2):89-101 [PubMed PMID: 742440]
Pudenz RH, Foltz EL. Hydrocephalus: overdrainage by ventricular shunts. A review and recommendations. Surgical neurology. 1991 Mar:35(3):200-12 [PubMed PMID: 1996449]
Bryant JR, Mantilla-Rivas E, Keating RF, Rana MS, Manrique M, Oh AK, Magge SN, Murnick J, Oluigbo CO, Myseros JS, Rogers GF. Craniosynostosis Develops in Half of Infants Treated for Hydrocephalus with a Ventriculoperitoneal Shunt. Plastic and reconstructive surgery. 2021 Jun 1:147(6):1390-1399. doi: 10.1097/PRS.0000000000007988. Epub [PubMed PMID: 34019511]
Kajdic N, Spazzapan P, Velnar T. Craniosynostosis - Recognition, clinical characteristics, and treatment. Bosnian journal of basic medical sciences. 2018 May 20:18(2):110-116. doi: 10.17305/bjbms.2017.2083. Epub 2018 May 20 [PubMed PMID: 28623672]
Blessing M,Gallagher ER, Epidemiology, Genetics, and Pathophysiology of Craniosynostosis. Oral and maxillofacial surgery clinics of North America. 2022 Aug; [PubMed PMID: 35787827]
Governale LS. Craniosynostosis. Pediatric neurology. 2015 Nov:53(5):394-401. doi: 10.1016/j.pediatrneurol.2015.07.006. Epub 2015 Jul 22 [PubMed PMID: 26371995]
Pfaff MJ, Fenton R, Mittal A, Mocharnuk JW, Owoc MS, Bruce MK, Beiriger JW, Losee JE, Goldstein JA. The Clinical Significance of Clinocephaly in Late-Presentation Sagittal Craniosynostosis. The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial Association. 2023 May:60(5):521-525. doi: 10.1177/10556656211064484. Epub 2022 May 11 [PubMed PMID: 35538850]
Fearon JA, Dimas V, Ditthakasem K. Lambdoid Craniosynostosis: The Relationship with Chiari Deformations and an Analysis of Surgical Outcomes. Plastic and reconstructive surgery. 2016 Mar:137(3):946-951. doi: 10.1097/01.prs.0000480014.18541.d8. Epub [PubMed PMID: 26910678]
Czorny A, Yettou H, Forlodou P, Stricker M. [The posterior part of the skull. Classification of dysmorphism. Original treatment: turned biparietal flap transposition]. Neuro-Chirurgie. 1995:41(4):295-314 [PubMed PMID: 8524442]
Manjila S, Chim H, Eisele S, Chowdhry SA, Gosain AK, Cohen AR. History of the Kleeblattschädel deformity: origin of concepts and evolution of management in the past 50 years. Neurosurgical focus. 2010 Dec:29(6):E7. doi: 10.3171/2010.9.FOCUS10212. Epub [PubMed PMID: 21121721]
Frassanito P, Palombi D, Tamburrini G. Craniosynostosis and hydrocephalus: relevance and treatment modalities. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2021 Nov:37(11):3465-3473. doi: 10.1007/s00381-021-05158-z. Epub 2021 Apr 7 [PubMed PMID: 33829280]
Collmann H, Sörensen N, Krauss J. Hydrocephalus in craniosynostosis: a review. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2005 Oct:21(10):902-12 [PubMed PMID: 15864600]
Bonfield CM, Shannon CN, Reeder RW, Browd S, Drake J, Hauptman JS, Kulkarni AV, Limbrick DD, McDonald PJ, Naftel R, Pollack IF, Riva-Cambrin J, Rozzelle C, Tamber MS, Whitehead WE, Kestle JRW, Wellons JC, Hydrocephalus Clinical Research Network (HCRN). Hydrocephalus treatment in patients with craniosynostosis: an analysis from the Hydrocephalus Clinical Research Network prospective registry. Neurosurgical focus. 2021 Apr:50(4):E11. doi: 10.3171/2021.1.FOCUS20979. Epub [PubMed PMID: 33794488]
Strahle J, Muraszko KM, Buchman SR, Kapurch J, Garton HJ, Maher CO. Chiari malformation associated with craniosynostosis. Neurosurgical focus. 2011 Sep:31(3):E2. doi: 10.3171/2011.6.FOCUS11107. Epub [PubMed PMID: 21882907]
Cinalli G, Spennato P, Sainte-Rose C, Arnaud E, Aliberti F, Brunelle F, Cianciulli E, Renier D. Chiari malformation in craniosynostosis. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2005 Oct:21(10):889-901 [PubMed PMID: 15875201]
Davis AA, Zuccoli G, Haredy MM, Runkel L, Losee J, Pollack IF, Tamber MS, Tyler-Kabara E, Goldstein JA, Nischal KK. The Incidence of Chiari Malformations in Patients with Isolated Sagittal Synostosis. Plastic and reconstructive surgery. Global open. 2019 Feb:7(2):e2090. doi: 10.1097/GOX.0000000000002090. Epub 2019 Feb 12 [PubMed PMID: 30881832]
Padmanabhan V, Hegde AM, Rai K. Crouzon's syndrome: A review of literature and case report. Contemporary clinical dentistry. 2011 Jul:2(3):211-4. doi: 10.4103/0976-237X.86464. Epub [PubMed PMID: 22215936]
Level 3 (low-level) evidenceAgochukwu NB, Solomon BD, Muenke M. Impact of genetics on the diagnosis and clinical management of syndromic craniosynostoses. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2012 Sep:28(9):1447-63. doi: 10.1007/s00381-012-1756-2. Epub 2012 Aug 8 [PubMed PMID: 22872262]
O'Hara J, Ruggiero F, Wilson L, James G, Glass G, Jeelani O, Ong J, Bowman R, Wyatt M, Evans R, Samuels M, Hayward R, Dunaway DJ. Syndromic Craniosynostosis: Complexities of Clinical Care. Molecular syndromology. 2019 Feb:10(1-2):83-97. doi: 10.1159/000495739. Epub 2019 Jan 16 [PubMed PMID: 30976282]
Das JM, Winters R. Pfeiffer Syndrome. StatPearls. 2025 Jan:(): [PubMed PMID: 30422477]
Massimi L, Bianchi F, Frassanito P, Calandrelli R, Tamburrini G, Caldarelli M. Imaging in craniosynostosis: when and what? Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2019 Nov:35(11):2055-2069. doi: 10.1007/s00381-019-04278-x. Epub 2019 Jul 9 [PubMed PMID: 31289853]
Senarath-Yapa K, Chung MT, McArdle A, Wong VW, Quarto N, Longaker MT, Wan DC. Craniosynostosis: molecular pathways and future pharmacologic therapy. Organogenesis. 2012 Oct-Dec:8(4):103-13. doi: 10.4161/org.23307. Epub 2012 Oct 1 [PubMed PMID: 23249483]
Level 3 (low-level) evidenceAbu-Sittah GS, Jeelani O, Dunaway D, Hayward R. Raised intracranial pressure in Crouzon syndrome: incidence, causes, and management. Journal of neurosurgery. Pediatrics. 2016 Apr:17(4):469-75. doi: 10.3171/2015.6.PEDS15177. Epub 2015 Nov 27 [PubMed PMID: 26613275]
Marucci DD, Dunaway DJ, Jones BM, Hayward RD. Raised intracranial pressure in Apert syndrome. Plastic and reconstructive surgery. 2008 Oct:122(4):1162-1168. doi: 10.1097/PRS.0b013e31818458f0. Epub [PubMed PMID: 18827651]
Driessen C, Eveleens J, Bleyen I, van Veelen ML, Joosten K, Mathijssen I. Optical coherence tomography: a quantitative tool to screen for papilledema in craniosynostosis. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2014 Jun:30(6):1067-73. doi: 10.1007/s00381-014-2376-9. Epub 2014 Feb 12 [PubMed PMID: 24519451]
Swanson JW, Aleman TS, Xu W, Ying GS, Pan W, Liu GT, Lang SS, Heuer GG, Storm PB, Bartlett SP, Katowitz WR, Taylor JA. Evaluation of Optical Coherence Tomography to Detect Elevated Intracranial Pressure in Children. JAMA ophthalmology. 2017 Apr 1:135(4):320-328. doi: 10.1001/jamaophthalmol.2017.0025. Epub [PubMed PMID: 28241164]
Tamburrini G, Caldarelli M, Massimi L, Santini P, Di Rocco C. Intracranial pressure monitoring in children with single suture and complex craniosynostosis: a review. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2005 Oct:21(10):913-21 [PubMed PMID: 15871027]
Al-Saleh S, Riekstins A, Forrest CR, Philips JH, Gibbons J, Narang I. Sleep-related disordered breathing in children with syndromic craniosynostosis. Journal of cranio-maxillo-facial surgery : official publication of the European Association for Cranio-Maxillo-Facial Surgery. 2011 Apr:39(3):153-7. doi: 10.1016/j.jcms.2010.04.011. Epub 2010 Jun 2 [PubMed PMID: 20627744]
Tan HL, Kheirandish-Gozal L, Abel F, Gozal D. Craniofacial syndromes and sleep-related breathing disorders. Sleep medicine reviews. 2016 Jun:27():74-88. doi: 10.1016/j.smrv.2015.05.010. Epub 2015 Jun 6 [PubMed PMID: 26454241]
Prager JD, Wang EW, Molter DW. Hearing loss in pediatric patients with isolated nonsyndromic sagittal synostosis. International journal of pediatric otorhinolaryngology. 2008 Feb:72(2):223-7 [PubMed PMID: 18053583]
Agochukwu NB, Solomon BD, Muenke M. Hearing loss in syndromic craniosynostoses: otologic manifestations and clinical findings. International journal of pediatric otorhinolaryngology. 2014 Dec:78(12):2037-47. doi: 10.1016/j.ijporl.2014.09.019. Epub 2014 Sep 28 [PubMed PMID: 25441602]
Melin AA, Moffitt J, Hopkins DC, Shah MN, Fletcher SA, Sandberg DI, Teichgraeber JF, Greives MR. Is Less Actually More? An Evaluation of Surgical Outcomes Between Endoscopic Suturectomy and Open Cranial Vault Remodeling for Craniosynostosis. The Journal of craniofacial surgery. 2020 Jun:31(4):924-926. doi: 10.1097/SCS.0000000000006152. Epub [PubMed PMID: 32049919]
Arko L 4th, Swanson JW, Fierst TM, Henn RE, Chang D, Storm PB, Bartlett SP, Taylor JA, Heuer GG. Spring-mediated sagittal craniosynostosis treatment at the Children's Hospital of Philadelphia: technical notes and literature review. Neurosurgical focus. 2015 May:38(5):E7. doi: 10.3171/2015.3.FOCUS153. Epub [PubMed PMID: 25929969]
Kalmar CL, Swanson JW, Shakir S, Tucker AM, Kennedy BC, Storm PB, Heuer GG, Bartlett SP, Taylor JA, Lang SS. Spring-mediated cranioplasty for sagittal craniosynostosis. Neurosurgical focus: Video. 2021 Apr:4(2):V6. doi: 10.3171/2021.1.FOCVID2060. Epub 2021 Apr 1 [PubMed PMID: 36284840]
Linz C, Kunz F, Böhm H, Schweitzer T. Positional Skull Deformities. Deutsches Arzteblatt international. 2017 Aug 7:114(31-32):535-542. doi: 10.3238/arztebl.2017.0535. Epub [PubMed PMID: 28835328]
McKee RM, Kamel GN, Cronin BJ, Ewing E, Lance SH, Gosman AA. A Comparison of Intracranial Volumes and Metopic Index in Patients With Isolated Metopic Ridge, Metopic Craniosynostosis, and Normal Healthy Children. The Journal of craniofacial surgery. 2021 Jan-Feb 01:32(1):108-112. doi: 10.1097/SCS.0000000000007044. Epub [PubMed PMID: 33186289]
Cho MJ,Kane AA,Seaward JR,Hallac RR, Metopic [PubMed PMID: 27449480]
Birgfeld CB, Saltzman BS, Hing AV, Heike CL, Khanna PC, Gruss JS, Hopper RA. Making the diagnosis: metopic ridge versus metopic craniosynostosis. The Journal of craniofacial surgery. 2013 Jan:24(1):178-85. doi: 10.1097/SCS.0b013e31826683d1. Epub [PubMed PMID: 23348281]
Level 2 (mid-level) evidenceStellwagen L, Hubbard E, Chambers C, Jones KL. Torticollis, facial asymmetry and plagiocephaly in normal newborns. Archives of disease in childhood. 2008 Oct:93(10):827-31. doi: 10.1136/adc.2007.124123. Epub 2008 Apr 1 [PubMed PMID: 18381343]
Hollier L, Kim J, Grayson BH, McCarthy JG. Congenital muscular torticollis and the associated craniofacial changes. Plastic and reconstructive surgery. 2000 Mar:105(3):827-35 [PubMed PMID: 10724239]
García-Mato D, Ochandiano S, García-Sevilla M, Navarro-Cuéllar C, Darriba-Allés JV, García-Leal R, Calvo-Haro JA, Pérez-Mañanes R, Salmerón JI, Pascau J. Craniosynostosis surgery: workflow based on virtual surgical planning, intraoperative navigation and 3D printed patient-specific guides and templates. Scientific reports. 2019 Nov 27:9(1):17691. doi: 10.1038/s41598-019-54148-4. Epub 2019 Nov 27 [PubMed PMID: 31776390]
Mardini S, Alsubaie S, Cayci C, Chim H, Wetjen N. Three-dimensional preoperative virtual planning and template use for surgical correction of craniosynostosis. Journal of plastic, reconstructive & aesthetic surgery : JPRAS. 2014 Mar:67(3):336-43. doi: 10.1016/j.bjps.2013.11.004. Epub 2013 Nov 21 [PubMed PMID: 24333232]
Seruya M, Borsuk DE, Khalifian S, Carson BS, Dalesio NM, Dorafshar AH. Computer-aided design and manufacturing in craniosynostosis surgery. The Journal of craniofacial surgery. 2013 Jul:24(4):1100-5. doi: 10.1097/SCS.0b013e31828b7021. Epub [PubMed PMID: 23851748]
Kapp-Simon KA,Speltz ML,Cunningham ML,Patel PK,Tomita T, Neurodevelopment of children with single suture craniosynostosis: a review. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2007 Mar; [PubMed PMID: 17186250]
Gabrick KS,Wu RT,Singh A,Persing JA,Alperovich M, Radiographic Severity of Metopic Craniosynostosis Correlates with Long-Term Neurocognitive Outcomes. Plastic and reconstructive surgery. 2020 May; [PubMed PMID: 32332546]
Almeida MN, Alper DP, Parikh N, De Baun H, Kammien A, Persing JA, Alperovich M. Comparison of emotional and behavioral regulation between metopic and sagittal synostosis. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2024 Sep:40(9):2789-2799. doi: 10.1007/s00381-024-06387-8. Epub 2024 May 1 [PubMed PMID: 38691155]
Bellew M, Chumas P. Long-term developmental follow-up in children with nonsyndromic craniosynostosis. Journal of neurosurgery. Pediatrics. 2015 Oct:16(4):445-51. doi: 10.3171/2015.3.PEDS14567. Epub 2015 Jul 24 [PubMed PMID: 26207667]
Starr JR, Collett BR, Gaither R, Kapp-Simon KA, Cradock MM, Cunningham ML, Speltz ML. Multicenter study of neurodevelopment in 3-year-old children with and without single-suture craniosynostosis. Archives of pediatrics & adolescent medicine. 2012 Jun 1:166(6):536-42. doi: 10.1001/archpediatrics.2011.1800. Epub [PubMed PMID: 22312170]
Level 2 (mid-level) evidenceVaca EE, Sheth N, Purnell CA, McGrath JL, Gosain AK. Secondary Suture Fusion after Primary Correction of Nonsyndromic Craniosynostosis: Recognition of the Problem and Identification of Risk Factors. Plastic and reconstructive surgery. 2020 Feb:145(2):493-503. doi: 10.1097/PRS.0000000000006491. Epub [PubMed PMID: 31985646]
Shim KW, Park EK, Kim JS, Kim YO, Kim DS. Neurodevelopmental Problems in Non-Syndromic Craniosynostosis. Journal of Korean Neurosurgical Society. 2016 May:59(3):242-6. doi: 10.3340/jkns.2016.59.3.242. Epub 2016 May 10 [PubMed PMID: 27226855]
Han RH, Nguyen DC, Bruck BS, Skolnick GB, Yarbrough CK, Naidoo SD, Patel KB, Kane AA, Woo AS, Smyth MD. Characterization of complications associated with open and endoscopic craniosynostosis surgery at a single institution. Journal of neurosurgery. Pediatrics. 2016 Mar:17(3):361-70. doi: 10.3171/2015.7.PEDS15187. Epub 2015 Nov 20 [PubMed PMID: 26588461]