Introduction
Hypoplastic left heart syndrome (HLHS) is a congenital heart defect characterized by the underdevelopment of the left-sided structures of the heart. This includes the mitral valve, left ventricle (LV), aortic valve, ascending aorta, and aortic arch. HLHS was first described in 1952 by Lev, and in 1958, Nadas and Noonan introduced it as a syndrome, referring to HLHS as a combined aortic and mitral atresia malformation.[1]
HLHS occurs in approximately 1 in 5000 newborns, making up about 3% of all infants born with congenital heart disease. Although rare, HLHS accounts for 23% of all cardiac-related deaths in the first week of life. Infants born with HLHS depend on a patent ductus arteriosus and interatrial communication for survival until they can undergo surgical intervention. A continuous infusion of prostaglandin E1 is necessary to maintain ductal patency.[2][3]
Before the 1980s, infants with this condition received comfort care and typically did not survive long after birth. However, due to significant medical and surgical advancements, current treatment options include a staged surgical approach that may start with the Norwood or Sano procedure during the neonatal period. Alternatively, a hybrid procedure, which combines cardiac catheterization with surgery off cardiopulmonary bypass or heart transplantation, may be considered.
Fetal cardiac interventions are available in certain cases at select centers, although they carry a high mortality risk. Palliative care is also an option for these infants. If the infant survives the first stage of surgical or hybrid treatment (ie, the Norwood/hybrid), 2 palliative surgical operations typically follow: the hemi-Fontan or bidirectional Glenn shunt, followed by the Fontan procedures.[1]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
HLHS is a complex congenital heart defect characterized by underdevelopment of the left-sided cardiac structures, including the LV, mitral valve, aortic valve, and ascending aorta. These anomalies prevent the left heart from adequately pumping oxygenated blood to the systemic circulation, necessitating immediate intervention after birth to sustain life. The etiology of HLHS is multifactorial, involving a combination of genetic, environmental, and possibly epigenetic influences.
Genetic Contributions
HLHS has been associated with chromosomal abnormalities and single-gene mutations. Specific genetic syndromes linked to HLHS include Turner syndrome, trisomy 13, and trisomy 18. Mutations in genes critical for cardiac development, such as those in the Notch signaling pathway (eg, NOTCH1), have also been implicated. Familial clustering suggests a hereditary component, with HLHS exhibiting autosomal dominant inheritance with variable expressivity in some cases. Genome-wide studies have identified additional candidate loci, though the precise genetic underpinnings remain incomplete.
Developmental Pathophysiology
The primary defect in HLHS arises during embryogenesis, between weeks 4 and 8 of gestation, due to abnormal development of the left-sided heart structures. The main factors leading to this defect are atresia or critical stenosis of the aortic or mitral valves, resulting in hypoplasia of the LV, ascending aorta, and aortic arch. Other etiologies include restricted LV inflow caused by a restrictive foramen ovale or leftward posterior deviation of the septum primum, generating mitral valve stenosis or atresia.[3] Insufficient blood flow through the left heart during fetal development contributes to hypoplasia, while intrinsic genetic defects may impair cellular proliferation, differentiation, or signaling pathways critical for normal heart morphogenesis.
Environmental and Maternal Factors
While specific environmental exposures have not been definitively linked to HLHS, maternal factors such as pregestational diabetes, advanced maternal age, and certain teratogenic exposures may modestly increase the risk. Nutritional deficiencies (ie, low folate levels) and maternal obesity have also been associated with congenital heart defects, though their direct role in HLHS remains speculative.
Epidemiology
HLHS is a rare congenital heart defect, accounting for approximately 2% to 3% of all congenital heart defects and about 6% to 9% of critical congenital heart disease requiring surgery or catheter-based intervention in the neonatal period. The incidence of HLHS is estimated to be 0.16 to 0.36 per 1000 live births and accounts for 1.4% to 3.8% of all congenital heart defects.[2] Males are affected more frequently than females, with a male-to-female ratio of approximately 1.5:1. HLHS is responsible for 23% of cardiac deaths during the first week of life, highlighting its significant contribution to early neonatal mortality.[1]
Several study results have suggested a multifactorial etiology involving genetic and environmental factors. Although most cases of HLHS are sporadic, up to 20% may occur in association with chromosomal abnormalities or syndromes, such as Turner syndrome, trisomy 13, or trisomy 18. Familial clustering has been observed, and mutations in genes like NOTCH1 and HAND1 have been implicated, suggesting a genetic predisposition in some cases.
HLHS is often diagnosed prenatally through routine fetal ultrasonography, with advances in imaging techniques enabling earlier and more accurate detection. Early diagnosis has improved perinatal management and outcomes. Despite its rarity, HLHS remains a leading cause of neonatal mortality due to congenital heart disease if left untreated, underscoring the importance of early intervention and specialized care.
Pathophysiology
HLHS is a critical congenital heart defect resulting from the underdevelopment of the left-sided cardiac structures, including the LV, mitral valve, aortic valve, and ascending aorta. The condition arises from multifactorial etiologies, with the main factor being an obstruction to LV outflow caused by aortic stenosis or atresia during fetal development. This obstruction increases LV afterload, causing hypertrophy and subsequently inhibiting ventricular growth, resulting in hypoplasia. Reduced LV blood flow further impairs its development and diminishes blood flow to the ascending aorta and aortic arch. Increased left atrial pressure due to inadequate LV egress causes bidirectional flow or reversal through the foramen ovale, exacerbating hypoplasia. Mitral stenosis or atresia can similarly limit LV inflow, leading to decreased preload, reduced pressure, and underdevelopment of the LV and associated structures.
Neonates with HLHS are entirely dependent on a patent ductus arteriosus (PDA) to sustain systemic and coronary perfusion. These patients are considered to have parallel systemic and pulmonary circulations, unlike the normal heart's series circulation.[3] Blood from the right ventricle is ejected through the pulmonary valve into the pulmonary artery, which flows into the pulmonary circulation and retrograde through the PDA to supply the systemic circulation, including the coronary arteries. The left atrium serves only as a conduit for pulmonary venous blood to reach the right atrium through interatrial communication, such as a patent foramen ovale or atrial septal defect. Restricted interatrial communication leads to elevated left atrial pressure, pulmonary venous congestion, and compromised oxygenation, often requiring emergent balloon atrial septostomy.
Pulmonary and systemic blood flow balance depends on the ratio of pulmonary vascular resistance (PVR) to systemic vascular resistance. High PVR favors systemic circulation at birth, but as PVR decreases postnatally, pulmonary blood flow increases, compromising systemic perfusion.[4][5] Medical or surgical interventions aim to maintain an optimal pulmonary and systemic circulation balance, ideally at a 1:1 ratio.[6] Closure of the PDA results in severe systemic hypoperfusion and rapid cardiovascular collapse, necessitating continuous infusion of prostaglandin E1 to maintain ductal patency.[3][4][7][8]
In HLHS, the nonfunctional LV is bypassed entirely. Oxygenated pulmonary venous blood mixes with deoxygenated systemic venous blood in the right atrium, resulting in systemic oxygen saturation levels of 75% to 85%, causing cyanosis. Over time, the right ventricle is subject to increased workload, and without staged surgical palliation, it cannot sustain systemic and pulmonary circulation long-term. Neonates with a restrictive foramen ovale or intact atrial septum may require an emergent percutaneous balloon atrial septostomy in the cardiac catheterization lab after birth to create a larger interatrial communication.[9] Early recognition, stabilization, and a staged surgical approach are critical to improving survival and outcomes in neonates with HLHS.
History and Physical
History
Neonates with HLHS are often born slightly premature, typically between 37 and 38 weeks of gestation.[4] While prenatal diagnosis via fetal ultrasound is possible, undiagnosed cases may appear normal at birth due to a PDA during the first 24 to 48 hours. Parents may report signs such as feeding difficulties, lethargy, or cyanosis within the first few days of life as the ductus arteriosus begins to close.
Physical Findings
Initial examination may not reveal a heart murmur, as the condition depends on the ductus arteriosus for systemic perfusion. Early findings include cyanosis, tachypnea, and oxygen saturations less than 95% due to mixing of oxygenated and deoxygenated blood, prompting further evaluation with echocardiography.[10][11] If the ductus arteriosus closes, symptoms of compromised systemic perfusion arise, including poor perfusion, tachycardia, hypotension, pallor, and weak peripheral pulses. Without immediate intervention, patients develop cardiogenic shock characterized by metabolic acidosis, lethargy, oliguria or anuria, and signs of multiorgan dysfunction.
Evaluation
The evaluation of HLHS relies on a combination of imaging modalities, laboratory tests, and genetic assessments to confirm the diagnosis, plan management, and guide surgical decision-making.
Laboratory and Genetic Testing
Comprehensive blood work aids in assessing systemic and metabolic stability in neonates with HLHS. An arterial blood gas evaluates oxygenation, ventilation, and acid-base balance, while a complete blood count detects anemia or polycythemia, which can exacerbate cyanosis. Electrolyte panels and renal function tests monitor organ perfusion and detect metabolic derangements. Genetic testing is essential to identify associated chromosomal abnormalities, such as Turner syndrome, DiGeorge syndrome, or Down syndrome, as these neonates have higher morbidity and mortality rates as well as experience longer hospital stays after surgery.[12]
Imaging Evaluation
Echocardiography is the gold standard for diagnosing HLHS, and it can detect the condition prenatally or after birth. Prenatal echocardiography may reveal progressive LV hypoplasia and hypoplasia of the aortic arch, though these changes might not be fully apparent until later in gestation.[13] The prenatal diagnosis rates vary from 39% to 75%.[4] Echocardiography will reveal a small LV, a small mitral/aortic valve, and a small ascending aorta and aortic arch. The right ventricle (RV) and right atrium (RA) often appear dilated from the increased volume load during the prenatal period.[13] A 4-chamber view is ideal for comparing the size of both ventricles. Echocardiography is also used to evaluate the flow through the foramen ovale and the potential need for emergent cardiac intervention, such as a percutaneous balloon atrial septostomy immediately after birth.[1]
Additional imaging tools enhance diagnostic precision and surgical planning. Chest x-rays typically reveal an enlarged cardiac silhouette and pulmonary venous hypertension, serving as an initial diagnostic tool but offering limited structural detail.[14] Magnetic resonance imaging/angiography (MRI/MRA) is critical in complex cases, particularly when the LV is mildly hypoplastic, and 2-ventricle surgical repair is considered, either during the neonatal period or following an initial single-ventricle palliation.[15][16] MRA quantifies left and RV volumes and evaluates mitral versus tricuspid valve inflows to determine the optimal surgical approach. Following initial single-ventricle palliation, MRA is increasingly used to assess systemic RV systolic function, detect tricuspid regurgitation, and identify complications such as residual coarctation or pulmonary artery stenosis before the second-stage bidirectional cavopulmonary anastomosis.[17][18]
Cardiac computed tomography angiography (CTA) offers a faster alternative to MRI for preoperative assessment, providing detailed anatomic insights at the expense of radiation exposure (see Video. Hypoplastic Aorta in Hypoplastic Left Heart Syndrome).[19] After the third-stage Fontan procedure, MRI complements echocardiography, particularly in older patients with poor acoustic windows, by enhancing risk stratification and supporting transplant-free survival predictions.[20] Additionally, neonates with HLHS face a heightened risk of cerebral intraventricular hemorrhage, necessitating head ultrasounds before surgical repair to ensure safety and guide interventions.[21]
Treatment / Management
Prenatal Management
HLHS is one of the most commonly identified congenital cardiac lesions during routine fetal ultrasound screenings, necessitating sensitive and comprehensive parental counseling.[13] Parents must be educated about the available treatment options, including the 3-stage surgical repair or comfort care after birth. These discussions should involve cardiac specialists and social workers who can provide resources such as family support groups. Elective termination rates following an HLHS diagnosis vary widely, ranging from 12% to 48%, highlighting the need for empathetic, patient-centered counseling tailored to individual circumstances.[4]
For families choosing the 3-stage surgical approach, close monitoring with serial fetal cardiac exams and obstetrical care near a pediatric cardiac center is essential. An interprofessional care team must coordinate a detailed birth plan, including obstetricians, cardiologists, cardiac surgeons, neonatologists, and anesthesiologists. This preparation may include readiness for emergent procedures such as atrial septostomy or extracorporeal membrane oxygenation cannulation immediately after delivery. Despite the higher risk of prematurity, data suggest that delivery should ideally occur at 39 weeks of gestation unless clinical indications necessitate earlier delivery.[4] Prenatal diagnosis enables better planning, but the timing and location of delivery remain critical for optimizing neonatal outcomes.
Postnatal Management
Neonates with HLHS are critically ill and will need to be managed in the intensive care unit and stabilized before surgical intervention during the first week of life. Initial management will include:
- Maintaining ductal patency
- Neonates suspected of having HLHS, identified either prenatally or postnatally, should promptly undergo a transthoracic echocardiogram to confirm the diagnosis. Once HLHS is verified, an intravenous prostaglandin E1 (PGE1) infusion must be initiated immediately to maintain ductal patency, ensuring adequate systemic blood flow. The recommended initial dose of PGE1 is 0.05 to 0.1 mcg/kg/minute, which can be gradually reduced to 0.02 to 0.01 mcg/kg/minute once ductal patency is established. Using the lowest effective dose of PGE1 is crucial to minimize adverse effects, such as respiratory depression and hypotension, while maintaining systemic circulation stability.[7][22]
- Avoiding excess pulmonary blood flow
- Due to the decrease in PVR after birth, neonates with HLHS may experience significant systemic hypoperfusion. Maintaining a balance between the pulmonary and systemic circulations is critical. Supplemental oxygen should generally be avoided, as oxygen saturations in the 70s and 80s are acceptable in this population. If oxygen saturation levels rise too high, measures such as intubation, sedation, paralysis, and mechanical ventilation may be required to promote hypoventilation. This allows the artial pressure of carbon dioxide levels to be maintained from 45 to 50 mm Hg, which helps increase PVR.
- If these interventions are ineffective in raising PVR, ventilation with a fraction of inspired oxygen of 15% to 19%, achieved by using supplemental nitrogen or carbon dioxide, may be considered to enhance PVR further. This approach helps optimize systemic blood flow, preventing hypoperfusion and the associated metabolic acidosis. Any metabolic acidosis should be corrected with sodium bicarbonate. Maintaining a hematocrit level from 40% to 45% also ensures optimal oxygen-carrying capacity to support the neonate's circulatory needs.[7]
- Ensuring adequate blood flow from the left to the RA
- During the echocardiogram for neonates suspected of having HLHS, the atrial septum will be carefully evaluated. In many cases, a catheter-based or surgical septostomy is required shortly after birth to relieve obstruction to left atrial flow. This procedure creates a passage in the atrial septum, allowing decompression of the left atrium into the RA. This adjustment enables blood flow into the RV, then through the PA, and into the aorta via a PDA, maintaining systemic circulation.[23]
- If a restrictive foramen ovale or an intact atrial septum is identified, the newborn will be urgently transferred to the cardiac catheterization laboratory. The procedure focuses on enlarging the interatrial communication to decompress the left atrium. Failure to address a restrictive or intact septum can lead to an irreversible increase in PVR, significantly increasing mortality and morbidity rates. Several percutaneous catheter-based techniques can be employed, including the Rashkind balloon atrial septostomy, static balloon septal dilation, and Park blade septostomy. In rare cases, an interatrial stent may be necessary to alleviate severe restriction and improve hemodynamic status.[7][9]
(A1)
Palliative Surgical Management
Survival for neonates with HLHS depends on a series of 3 palliative surgeries aimed at establishing a Fontan-type physiology, which separates systemic and pulmonary circulations. In this physiology, the RV assumes the role of the systemic ventricle, pumping oxygenated blood to the body while deoxygenated blood flows passively into the pulmonary circulation. The first stage, performed within the first week of life, ensures initial stabilization. The second stage occurs around 4 to 6 months, and the third stage is typically completed around age 2.[24]
While advancements in surgical, transcatheter, and pharmacological therapies have transformed HLHS from a fatal condition to a manageable one, long-term morbidity and mortality remain significant. These patients often face challenges, including heart failure, arrhythmias, and complications related to the Fontan circulation, necessitating lifelong specialized care and monitoring.[25]
Stage 1
- Norwood
- The stage 1 Norwood procedure, first described by Norwood et al in 1979, is the foundational surgical intervention for neonates with HLHS. This procedure allows the RV to assume the role of the systemic ventricle, with pulmonary blood flow supplied via a modified Blalock-Taussig (BT) shunt. To perform the Norwood procedure, surgeons use deep hypothermic circulatory arrest to reconstruct the aortic arch.[1]
- Key components of the procedure include:
- Atrial septum removal
- The removal facilitates the unimpeded flow of oxygenated blood from the left atrium and pulmonary veins into the RV.
- Neoaorta creation
- The hypoplastic ascending aorta is anastomosed to the main PA to establish a single outflow tract for systemic circulation, which is powered by the RV.
- Systemic-to-pulmonary shunt (BT shunt)
- The right subclavian or innominate artery is connected to the right PA, providing controlled pulmonary blood flow. The shunt’s size is tailored to balance pulmonary and systemic circulation, avoiding overcirculation.[1]
- Atrial septum removal
- This intricate procedure is pivotal in stabilizing neonates with HLHS and preparing them for subsequent staged surgeries.
- Sano
- The Sano procedure, introduced in the late 1990s, modifies the stage 1 Norwood procedure by replacing the BT shunt with a nonvalved RV-to-PA (pulmonary artery) conduit for pulmonary blood flow. While creating a neoaorta remains consistent with the original Norwood technique, the Sano procedure offers advantages such as higher diastolic pressures and improved coronary perfusion. However, its disadvantages include an increased risk of RV arrhythmias and potential ventricular impairment due to the incision required for conduit placement.[1]
- The Single Ventricle Reconstruction Trial, designed to compare the BT shunt and the RV-PA (Sano) shunt in neonates with HLHS, provided results with key insights:
- Short-term outcomes
- At 1 year, the Sano group demonstrated superior transplant-free survival.
- Intermediate outcomes
- By 6 years, differences in survival between the 2 groups were no longer evident.
- Long-term outcomes
- At 12 years postsurgery, shunt type had minimal impact on RV ejection fraction, maximal oxygen consumption, complication rates, or transplant-free survival. However, the Sano group showed higher incidences of protein-losing enteropathy and required more interventional catheterizations, while other morbidity rates were comparable.[25]
- Short-term outcomes
- These findings underscore the nuanced trade-offs between the 2 approaches, emphasizing the need for individualized patient care strategies.
- Hybrid
- The hybrid procedure offers an alternative to the Norwood procedure for neonates with HLHS who are high-risk surgical candidates, such as those with prematurity, low birth weight (<2 kg), or significant comorbidities.[26] Developed by Gibbs et al in 1993, this approach combines cardiac catheterization and an off-cardiopulmonary bypass surgical procedure.[1] The hybrid procedure minimizes physiologic stress by avoiding cardiopulmonary bypass and has been associated with a reduced 30-day mortality rate compared to the Norwood procedure in this population.
- The hybrid procedure involves:
- Bilateral pulmonary banding
- This is performed via sternotomy and restricts pulmonary blood flow to prevent pulmonary overcirculation and maintain systemic perfusion.[3]
- Ductus arteriosus stenting
- This is performed during cardiac catheterization and ensures systemic perfusion without requiring PGE1.
- Atrial septal intervention
- Stenting or balloon septostomy is completed via cardiac catheterization to ensure adequate interatrial communication.[1]
- Bilateral pulmonary banding
- Following the procedure, patients maintain a parallel circulation, requiring careful balancing of pulmonary and systemic blood flow to achieve a 1:1 ratio. Systemic oxygen saturations typically range from 75% to 85% due to the mixing of oxygenated and deoxygenated blood. Postoperative management is challenging, with a potential need for extracorporeal membrane oxygenation support. Despite advances, interstage mortality rates remain at 5% to 15%, emphasizing the complexity of care for these critically ill neonates.[7]
Interstage Mortality
When the Norwood procedure was initially introduced for newborns with HLHS, it was followed by the Fontan operation in a 2-stage palliative strategy. However, high mortality rates associated with the Fontan procedure prompted the development of a 3-stage surgical approach in 1988. This updated method incorporated the superior cavopulmonary connection as an intermediate step between the Norwood and Fontan procedures to improve outcomes.
Despite advancements, those with HLHS still face significant mortality risks during the interstage period between the Norwood procedure and the superior cavopulmonary connection, with rates ranging from 5% to 20%. Fortunately, this represents a considerable decrease compared to rates before 2011, largely due to several key improvements. Performing the superior cavopulmonary connection at a younger age has decreased the vulnerability of infants during this critical time. The introduction of interstage monitoring programs, where specialized healthcare teams meticulously observe infants in collaboration with their families, has further enhanced survival rates. The increased use of the RV-to-PA shunt has also contributed to better hemodynamic stability during this period.[27] Together, these measures have significantly improved the care and outcomes for infants with HLHS.
Stage 2
The hemi-Fontan and bidirectional Glenn procedures are vital second-stage surgeries in the 3-stage palliation for HLHS.[24] These procedures establish a superior cavopulmonary connection, redirecting systemic venous return from the upper body into the pulmonary circulation. This alteration reduces the RV's volume load, helping prevent RV hypertrophy and potentially improving diastolic function.[4]
Cardiac catheterization is performed to evaluate PA pressures before performing the hemi-Fontan or bidirectional Glenn procedures. Elevated pressures indicate increased PVR, which must be managed before proceeding to avoid postoperative complications. If pulmonary hypertension is untreated, it may compromise the success of the superior cavopulmonary connection.
The second stage transitions the circulation from a parallel system to a partial in-series system.[3] This change improves the efficiency of oxygen delivery by reducing the mixing of oxygenated and deoxygenated blood. The components of the second stage repair include:
- Superior cavopulmonary anastomosis
- In the bidirectional Glenn procedure, the proximal superior vena cava (SVC) is disconnected from the RA and oversewn, while the distal SVC is anastomosed to the right PA.
- In the hemi-Fontan procedure, the SVC is connected to the central and branch pulmonary arteries. Flow to the RA is interrupted by closing the SVC-RA junction with an allograft patch.[4]
- Neoaorta and aortic arch reconstruction
- As established in the first-stage Norwood procedure, the neoaorta is further refined if necessary.
- Atrial septectomy
- This ensures unobstructed blood flow between the atria and accommodates the single-ventricle physiology.
- PA band removal and arterioplasty
- PA bands applied during earlier stages are removed, and pulmonary arterioplasty is performed if required to optimize flow dynamics.
- Ductus arteriosus/stent complex removal
- This step finalizes the transition to a superior cavopulmonary connection, ensuring ductal patency is no longer needed for systemic circulation.[28]
(B2)
The hemi-Fontan and bidirectional Glenn procedures alleviate the volume load on the single functional ventricle. This reduction minimizes the risk of RV hypertrophy and promotes more efficient systemic circulation. Altering the circulatory system to a partial in-series configuration improves oxygenation, preparing the patient for the third-stage Fontan procedure. These interventions also mark a critical step in reducing the long-term complications associated with HLHS.
Stage 3
The Fontan procedure is the final stage in the surgical palliation of single-ventricle congenital heart defects, such as HLHS. This procedure separates systemic and pulmonary circulations, improving physiology by reducing ventricular volume load and enhancing systemic oxygenation.[24] While there is no universally established age or weight for Fontan completion, most centers perform this procedure when a patient is aged
between 2 and 4 years, ensuring adequate PA development and low PVR.[29] Preoperative evaluations include cardiac catheterization to measure PVR and assess ventricular function and atrioventricular valve competency. The blood flow into the pulmonary arteries is passive, so the pressures in the pulmonary arteries must be normal for the Fontan procedure to succeed. Additionally, the atrioventricular valve must be functioning properly.[30]
The complete Fontan circulation is achieved through 1 of 2 configurations: an extracardiac tunnel or an intraatrial tunnel. Both approaches establish a connection between the inferior vena cava (IVC) and the right PA to facilitate passive pulmonary blood flow.[3] In the lateral tunnel Fontan, the IVC is connected to the right PA using an intraatrial conduit, which directs blood flow through the atrium. Conversely, the extracardiac Fontan involves a direct anastomosis of the SVC to the right PA and incorporates an extracardiac prosthetic conduit to connect the IVC to the right PA.
The extracardiac Fontan procedure offers distinct advantages, including avoiding myocardial ischemia during surgery, fewer suture lines within the RA, and eliminating foreign material within the atrium.[4] These features reduce the risk of atrial reentry arrhythmias, such as atrial flutter, making it a preferred option in many cases. Both approaches aim to achieve optimal systemic and pulmonary separation while minimizing long-term complications associated with single-ventricle physiology.
Since pulmonary blood flow is passive, the success of the Fontan procedure depends on maintaining normal PA pressures and low PVR, as well as ensuring atrioventricular valve competence for efficient systemic circulation. In some cases, a fenestration (a small 3-5 mm opening) is created between the Fontan conduit and the common atrium. This controlled right-to-left shunt can help maintain cardiac output during periods of elevated pulmonary pressures or resistance and prevent acute Fontan failure by reducing systemic venous pressure. However, fenestrations result in lower systemic oxygen saturation and increase the risk of paradoxical embolism, potentially causing cerebrovascular accidents. Fenestrations are often closed via a transcatheter approach to restore full circulatory separation when pulmonary resistance normalizes.[31]
Although the Fontan circulation improves oxygenation and reduces systemic venous pressure, long-term complications can arise, including PLE due to elevated systemic venous pressure, thromboembolism from altered flow dynamics and venous stasis, arrhythmias, particularly in patients with prior atrial modifications, and progressive single-ventricle dysfunction leading to heart failure. Despite these challenges, the Fontan procedure significantly enhances the quality of life and survival for patients with single-ventricle physiology, though vigilant, lifelong follow-up is essential to manage associated morbidities.
Heart Transplantation
Given the limited availability of donor hearts for newborns, the primary treatment for HLHS is a multistage palliative surgical approach.[7] Primary heart transplantation is generally reserved for newborns with HLHS who are deemed too high-risk to undergo the staged repair process.[32] Unfortunately, approximately 20% of newborns on the transplant waiting list die before receiving a suitable organ.[33] To improve survival rates for these high-risk patients, some medical centers have adopted the hybrid (stage 1) procedure as a bridge to transplantation, providing an interim solution to stabilize systemic and pulmonary circulations until a donor heart becomes available.[32]
Differential Diagnosis
Several diseases or conditions can mimic the presentation of HLHS, particularly in neonates with cyanosis, signs of poor perfusion, or congestive heart failure. Accurate differentiation is critical, as management strategies for these conditions differ significantly from HLHS. Below is a discussion of diseases or conditions that can be mistaken for HLHS:
- Critical aortic stenosis
- Severe aortic stenosis can cause LV hypertrophy and poor systemic perfusion, resembling HLHS. However, the LV is typically present in critical aortic stenosis and may be dilated rather than hypoplastic. Echocardiography can differentiate this condition by visualizing the LV size and function and assessing the degree of aortic valve obstruction.
- Coarctation of the aorta
- Coarctation of the aorta, especially with a large ventricular septal defect, can lead to cyanosis and shock as the ductus arteriosus begins to close. Because of systemic hypoperfusion, this condition may be confused with HLHS, but echocardiography will show a normally sized or hypertrophied LV and an obstruction localized to the aortic isthmus.
- Shone complex
- Shone complex is a rare congenital heart condition characterized by multiple levels of left-sided obstructive lesions, including a supravalvular mitral membrane, a parachute mitral valve with all chordae attached to a single papillary muscle, subaortic stenosis caused by a fibromuscular ridge below the aortic valve, and coarctation of the aorta impairing systemic blood flow. This condition may be mistaken for HLHS because both conditions involve reduced left-sided cardiac output and systemic hypoperfusion. Symptoms such as cyanosis, poor perfusion, and heart failure, as the ductus arteriosus closes, are common to both, but echocardiography can differentiate them by identifying the discrete levels of obstruction in the Shone complex versus the global underdevelopment of the left heart in HLHS.
- Interrupted aortic arch
- This condition can present with systemic hypoperfusion and shock, similar to HLHS. This condition involves complete discontinuity of the aortic arch, but echocardiography or advanced imaging will reveal a normal or mildly hypoplastic LV and the specific site of aortic interruption.[8]
- Total anomalous pulmonary venous return (TAPVR)
- In TAPVR with obstruction, neonates may present with cyanosis and signs of low cardiac output that mimic HLHS. However, TAPVR is characterized by abnormal pulmonary venous drainage into the RA or systemic veins, which can be identified on echocardiography. The left-sided structures are typically normal in size and morphology.
- Pulmonary atresia with intact ventricular septum (PA/IVS)
- PA/IVS may resemble HLHS due to ductal-dependent systemic perfusion and cyanosis. However, in PA/IVS, the LV is usually normal in size, and the obstruction is at the pulmonary valve rather than the aortic or mitral valves.
- Ebstein anomaly of the tricuspid valve
- Severe forms of Ebstein anomaly can cause cyanosis and right-sided heart failure, potentially mimicking HLHS. However, echocardiography will show significant apical displacement of the tricuspid valve with atrialization of the RV, distinguishing this condition from HLHS.
Prognosis
One-third of neonates with HLHS die before undergoing any palliative surgical intervention. The pre-Fontan mortality rate is approximately 70%, whereas post-Fontan mortality decreases to around 5%. Neonates with a single ventricle face a heightened risk of cardiac arrest (12.7%) and a mortality rate of 62.3%. Some centers report survival rates as high as 90% following stage 1 Norwood palliation, but only two-thirds of children with HLHS survive to age 5, with an annual mortality rate of approximately 1% among Fontan patients.[4] The 2019 Society of Thoracic Surgeons Congenital Heart Surgery Database reported stage 1 palliation mortality at 15%, hemi-Fontan/Glenn at 1.8%, and Fontan at 1.0%.[34]
The Aristotle score is a system used to predict the survival chances of patients with congenital heart disease. This system calculates the risk of early mortality and morbidity, such as the length of stay in the intensive care unit, while also considering the anticipated difficulty of the surgical technique. This scoring system evaluates the complexity of the surgical procedure and assigns additional points based on the patient's current health condition. Points may be added for various factors, including anatomical variations, respiratory failure, shock, prematurity, and low weight. A score of 20 or higher is associated with a high mortality rate during surgery, and such scores are often observed in patients undergoing the Norwood procedure. The Aristotle score is calculated before surgery to help counsel parents and provide them with realistic expectations about the procedure.[35]
Thirty-five years after the first Norwood operation, adults with hypoplastic left heart syndrome (HLHS) represent a small but growing population; these individuals frequently exhibit reduced aerobic capacity, which tends to decline progressively with age. Morbidity and mortality in this population are frequently associated with complications such as heart failure, PLE, significant thrombotic events, malignant arrhythmias, and, in some cases, the need for heart transplantation.[36]
Neurodevelopmental Issues
Children with complex congenital cardiac diseases, particularly those with HLHS, face a higher risk of developmental delays, disabilities, and behavioral issues. Fetuses with HLHS exhibit decreased brain volume and metabolism, partially due to diminished cerebral oxygen delivery and utilization.[4] Older infants and toddlers with HLHS often exhibit developmental delays, with Psychomotor Development Index scores consistently lower than Mental Development Index scores. A specific area of concern is visual-motor integration, which shows notable delays.
However, children with HLHS who have undergone staged Fontan palliation can achieve normal developmental outcomes. Nonetheless, they remain at significant risk for learning disorders, lower academic performance, and behavioral challenges. Various factors likely influence neurodevelopment in these children, including associated neurological malformations, genetic conditions, anatomical features, surgical techniques, and intra-operative perfusion methods.[37]
Complications
HLHS is associated with numerous complications, both in pre and postsurgical intervention. These complications often arise due to the unique physiology of single-ventricle circulation and the complex surgeries required for palliation.
Preoperative Complications
- Cardiogenic shock
- Neonates with HLHS are dependent on a PDA for systemic circulation. Closure of the PDA can lead to severe systemic hypoperfusion, metabolic acidosis, and shock.
- Cyanosis
- Due to the mixing of oxygenated and deoxygenated blood, systemic oxygen saturation is typically between 75% and 85%.
- Restrictive foramen ovale
- This leads to pulmonary venous congestion and decreased cardiac output, often requiring emergent atrial septostomy.
- Arrhythmias
- These result from structural abnormalities or conduction system compromise.
Postoperative Complications
The staged palliation approach for HLHS carries distinct postoperative risks for each surgical stage due to the unique physiological challenges and complexities involved. Below are the postoperative complications associated with each procedural stage:
- Stage 1: Norwood procedure
- The Norwood procedure establishes systemic circulation using the RV and includes a modified BT shunt or Sano shunt for pulmonary blood flow.
- BT shunt thrombosis
- Coronary steal syndrome
- Arrhythmias
- Respiratory failure
- Bleeding and infection
- Renal dysfunction (oliguria/anuria)[4]
- The Norwood procedure establishes systemic circulation using the RV and includes a modified BT shunt or Sano shunt for pulmonary blood flow.
- Stage 2: Hemi-Fontan or Glenn shunt
- This procedure redirects systemic venous return from the upper body to the pulmonary arteries, reducing the volume load on the single ventricle.
- Decreased pulmonary flow
- Arrhythmias
- Thromboembolic events
- This procedure redirects systemic venous return from the upper body to the pulmonary arteries, reducing the volume load on the single ventricle.
- Stage 3: Fontan procedure
- The Fontan procedure completes the separation of systemic and pulmonary circulations, relying on passive pulmonary blood flow.
Postoperative and Rehabilitation Care
Stage 1 Palliation (Norwood, Sano, Hybrid)
Following stage 1 palliation for HLHS, patients are critically ill and require intensive monitoring and support in a specialized cardiothoracic intensive care unit. Postoperative management aims to optimize hemodynamics, balance systemic and pulmonary circulations, and ensure adequate organ perfusion. These patients' systemic and pulmonary circulations share a single functional ventricle. Maintaining a balance in blood flow between these is essential to ensure adequate oxygen delivery to both systemic and pulmonary tissues while avoiding complications like coronary steal or hypoxemia.
- Hemodynamic goals
- Mean arterial pressure: 40 to 45 mm Hg
- pH: 7.4
- Partial pressure of carbon dioxide, pCO2: 40 mm Hg
- Partial pressure of oxygen: 40 mm Hg
- Hematocrit: 40%
- Systemic oxygen saturation: 75% to 85%
- Lactic acid: Normal
- Regulation of hemodynamic parameters
- The balance between PVR and SVR is critical. These parameters are adjusted through targeted therapies, mechanical ventilation strategies, and blood product transfusion to maintain optimal hemodynamics.
- Strategies to adjust SVR
- Increase SVR
- Vasopressin, norepinephrine, epinephrine (alpha-agonists)
- Dopamine at higher doses
- Decrease SVR
- Milrinone (phosphodiesterase inhibitor)
- Nitroprusside (vasodilator)
- Nicardipine (calcium channel blocker)
- Increase SVR
- Strategies to adjust PVR
- Increase PVR
- Reduce oxygen delivery: Decrease fraction of inspired oxygen (FiO2)
- Increase pCO2: Reduce ventilation or add inhaled CO2
- Decrease PVR
- Increase oxygenation: Increase FiO2
- Use inhaled nitric oxide to promote pulmonary vasodilation
- Increase PVR
- Strategies to adjust SVR
- Postoperative support
- Mechanical ventilation to optimize oxygenation and CO2 clearance
- Inotropic support to maintain cardiac output and systemic perfusion
- Continuous monitoring of arterial blood gases, lactate levels, and hemodynamic parameters to guide therapy adjustments
- The balance between PVR and SVR is critical. These parameters are adjusted through targeted therapies, mechanical ventilation strategies, and blood product transfusion to maintain optimal hemodynamics.
Stage 2 Palliation (Hemi-Fontan/Bidirectional Glenn)
PVR must remain normal to promote proper pulmonary blood flow and sustain cardiac output. These patients benefit from early extubation because positive pressure ventilation increases intrathoracic pressure, which can reduce passive pulmonary blood flow from the SVC to the pulmonary circulation. Encouraging spontaneous ventilation will enhance systemic venous return. The goals for the postoperative period include the following:
- Systemic oxygen saturation between 75% to 85%
- Modest hypercapnia (pCO2 from 35-45 mm Hg)
- This may improve cerebral perfusion and pulmonary blood flow, thereby increasing systemic oxygen saturation.
- Optimize mixed venous saturation [4]
Stage 3 Palliation (Fontan Completion)
Fontan physiology relies on the passive blood flow from the systemic circulation into the pulmonary circulation. Consequently, it is crucial to maintain adequate blood volume. Keeping intrathoracic pressure low promotes blood flow through the Fontan circuit and into the pulmonary arteries. Early extubation after surgery is highly desirable, as it minimizes the duration of positive pressure ventilation, reduces intrathoracic pressure, and subsequently increases cardiac output.[4]
Imaging Surveillance
MRA and CTA play crucial roles in the postoperative management of patients following Fontan completion. These imaging modalities provide detailed anatomical and functional information for assessing complications, optimizing clinical outcomes, and planning interventions in patients with Fontan circulation.
- MRA
- MRA is particularly valuable because it is noninvasive and avoids ionizing radiation, making it suitable for repeated evaluations in a pediatric population. MRA frequently assesses the Fontan pathway and pulmonary vasculature for obstructions, stenoses, or thrombi. Additionally, it provides high-resolution images of the extracardiac or intraatrial tunnel and can assess for narrowing or kinks that may impair blood flow.
- MRA is also instrumental in evaluating systemic and pulmonary venous connections for anomalies or obstructions, particularly in cases where fenestrations or surgical modifications may complicate flow dynamics. The ability of MRA to perform flow mapping allows for the quantification of pulmonary and systemic blood flow ratios, aiding in the evaluation of hemodynamic efficiency. Furthermore, MRA is used to assess ventricular function and myocardial fibrosis through late gadolinium enhancement, helping to monitor systemic ventricle performance and identify early signs of dysfunction (see Video. Completed Fontan on Cardiac Magnetic Resonance Angiography).
- CTA
- CTA is often employed when rapid imaging is necessary or when MRA is contraindicated, such as in patients with certain metallic implants or severe claustrophobia. CTA provides high-resolution, 3-dimensional images of the Fontan circulation, enabling detailed assessment of vascular structures (see Image. Completed Fontan on Cardiac Computed Tomography). This modality is particularly useful for identifying complications such as pulmonary artery stenosis, collateral vessels, or thrombotic obstructions.
- In patients who are postoperative from the Fontan procedure, CTA is also a preferred modality for detecting abnormalities in the pulmonary vasculature, including arteriovenous malformations or venovenous collaterals that may contribute to cyanosis or inefficiency in the Fontan circulation. While CTA exposes patients to ionizing radiation, the advent of low-dose protocols and faster scan times has reduced the associated risks, making it a viable option in select cases.
Failing Fontan
Late Fontan failure can develop gradually over the years. Patients who have undergone the Fontan procedure often live with suboptimal cardiac output throughout their lives, and they may not notice signs of a gradual decline in their functional status until the deterioration is quite advanced. Additionally, it is generally observed that individuals in the Fontan population tend to be shorter in height compared to the general population.[41] Exercise capacity in those with Fontan is lower than in other individuals with repaired congenital heart disease. Results from recent studies have shown a 1% to 3% annual decline in maximal oxygen consumption starting in adolescence. This may be related to chronotropic incompetence or abnormal pulmonary compliance with exercise, which correlates with the need for hospitalization.[42]
Resting oxygen saturation levels below 90% indicate the potential presence of right-to-left shunting, which can occur either through intracardiac pathways or due to venovenous collateral flow into the left atrium. Another possible cause could be pulmonary arteriovenous fistulae. Over time, the RV, which is not equipped to function effectively as a systemic ventricle, begins to fail. Fontan physiology creates a state of chronic systemic venous hypertension due to obstructed blood flow through the pulmonary vasculature, resulting in a decreased cardiac output. Additionally, chronotropic insufficiency stemming from sinus node dysfunction, predominant junctional rhythm, or atrioventricular block further exacerbates the physiological impairments commonly associated with failing Fontan physiology. The risk of both supraventricular and ventricular arrhythmias, including the potential for arrhythmic sudden death, complicates this clinical picture.
The physiological challenges tied to a failing Fontan circulation can result in complications from elevated RA pressures. These include ascites, Fontan-associated liver disease, cirrhosis, PLE, lymphatic dysfunction, and plastic bronchitis. The heart is particularly susceptible to long-term stressors such as chronic preload deprivation and increased SVR, which can lead to both systolic and diastolic dysfunction. For many patients facing failing Fontan physiology, heart transplantation or the use of a ventricular assist device may be the only viable treatment options available.[43]
Fontan-Associated Liver Disease
Surveillance biopsies have shown that nearly 100% of patients who have undergone the Fontan procedure develop clinically silent fibrosis by adolescence. Over time, an increasing number of these patients require combined heart-liver transplantation due to advanced liver disease, which may include conditions such as bridging fibrosis, cirrhosis, and hepatocellular carcinoma. Liver biopsy is the gold standard for diagnosing advanced fibrosis in Fontan-associated liver disease. However, concerns about potential complications, such as bleeding, may discourage routine use. In this context, MRI elastography emerges as a noninvasive method for measuring liver stiffness, which can be valuable for assessing and guiding management of these cases.[40]
Deterrence and Patient Education
Deterrence
Although HLHS cannot be entirely prevented due to its complex genetic and environmental origins, certain measures may help reduce associated risks. Preconception counseling is crucial for those with a personal or family history of congenital heart defects. Consulting a genetic counselor or maternal-fetal specialist before conception allows for risk assessment and guidance. Additionally, avoiding teratogenic substances, infections, and harmful environmental exposures during pregnancy is vital.
Prenatal care plays a critical role in managing HLHS. Routine ultrasounds, including fetal echocardiography, facilitate early detection, enabling advanced planning and specialized perinatal care. Pregnant individuals should also avoid teratogens such as alcohol, tobacco, and specific medications like angiotensin-converting enzyme inhibitors. Ensuring optimal maternal health through proper management of conditions like diabetes or hypertension and maintaining adequate folic acid intake during pregnancy may further reduce congenital anomalies. Advances in research and genetic screening may eventually lead to preventive strategies or targeted therapies.
Patient Education
Education is essential to helping families navigate the complexities of HLHS. Parents should receive clear, comprehensible explanations of the condition, including its anatomical and physiological implications. Visual aids like diagrams or videos can enhance understanding. Families should also be informed about the staged surgical approach—the Norwood, Glenn, and Fontan procedures—and the goals of each stage, emphasizing the importance of medical monitoring and addressing potential complications.
Discussions about prognosis should be transparent, including survival rates and risks of complications such as arrhythmias, heart failure, or developmental delays. Parents should also be informed about the possibility of heart transplantation as a future option. Daily care guidelines, such as monitoring oxygen saturation and recognizing signs of distress like cyanosis, poor feeding, or lethargy, are crucial. Families should be educated about the importance of medication adherence, addressing feeding challenges, and attending routine healthcare appointments.
Lifestyle modifications tailored to the child’s capacity should be encouraged, along with dietary recommendations to support growth and avoid complications like PLE. Emotional and psychosocial support for families is equally important, with resources for psychological support and congenital heart defect support groups readily available. Families should also prioritize mental health care for both the child and themselves to cope with the challenges of living with HLHS.
As children grow, transitioning to adult congenital heart disease specialists is essential. Adolescents should be educated about their condition and empowered to manage their health, including understanding the risks associated with Fontan circulation. Emergency preparedness is another critical component. Families should be trained to recognize and respond to emergencies such as hypoxemia, arrhythmias, or thrombotic events and have clear instructions for accessing emergency care tailored to the patient’s unique cardiac anatomy. By addressing these elements, education and prevention efforts can significantly enhance the quality of life and long-term outcomes for individuals with HLHS and their families.
Pearls and Other Issues
Those pregnant with Fontan circulation face an elevated risk of complications, including arrhythmias, heart failure, intrauterine growth restriction, preterm delivery, and miscarriage. Among these, postpartum hemorrhage is the most common obstetric complication.[44] Consequently, these patients require coordinated care from a multidisciplinary team with expertise in adult congenital heart disease and high-risk obstetrics. Regular cardiology follow-up is crucial to monitor cardiovascular stability throughout pregnancy.
For those previously on anticoagulation therapy, management decisions should be individualized. Those with a history of thromboembolic events or atrial arrhythmias may benefit from low molecular weight heparin to mitigate thrombotic risks. Oxygen therapy is reserved for cases of maternal decompensation or significant arterial desaturation. Labor presents additional challenges, especially for women with impaired cardiac function, heart failure, or arrhythmias, as it can be poorly tolerated. Vaginal delivery is generally preferred and is facilitated by shortening the second stage of labor with the assistance of regional anesthesia. Cesarean delivery is reserved for obstetric indications.
Fluid management in Fontan patients during pregnancy and labor is critical, as these individuals have unique hemodynamic challenges. Precise balance is required to avoid volume overload, which could precipitate heart failure and hypovolemia, which may compromise perfusion.[45] An individualized care plan that addresses these complexities helps optimize maternal and fetal outcomes.
Enhancing Healthcare Team Outcomes
Managing HLHS demands a multidisciplinary approach, where clinicians, nurses, pharmacists, and therapists collaborate to enhance patient-centered care, outcomes, safety, and team performance. Clinicians lead by developing evidence-based treatment plans, interpreting complex diagnostics, and managing complications like heart failure and arrhythmias. Nurses are critical in monitoring subtle condition changes, educating families, and providing emotional support, while pharmacists optimize medication regimens and address potential drug interactions. Therapists contribute by addressing developmental delays and supporting rehabilitation.
Effective care requires standardized protocols for screening and managing complications, tailored care plans that consider disease progression and psychosocial needs, and advanced interventions like mechanical support or transplantation when necessary. Interprofessional communication is key. Regular team meetings facilitate comprehensive planning while coordinated efforts ensure seamless transitions to adult congenital heart disease care and long-term monitoring. By prioritizing collaboration, structured strategies, and patient-centered approaches, the team can significantly improve outcomes and quality of life for individuals with HLHS.
Media
(Click Video to Play)
Hypoplastic Aorta in Hypoplastic Left Heart Syndrome. Cardiac computed tomography cine clip demonstrating the hypoplastic aorta.
Contributed by A Thomas, MD
(Click Image to Enlarge)
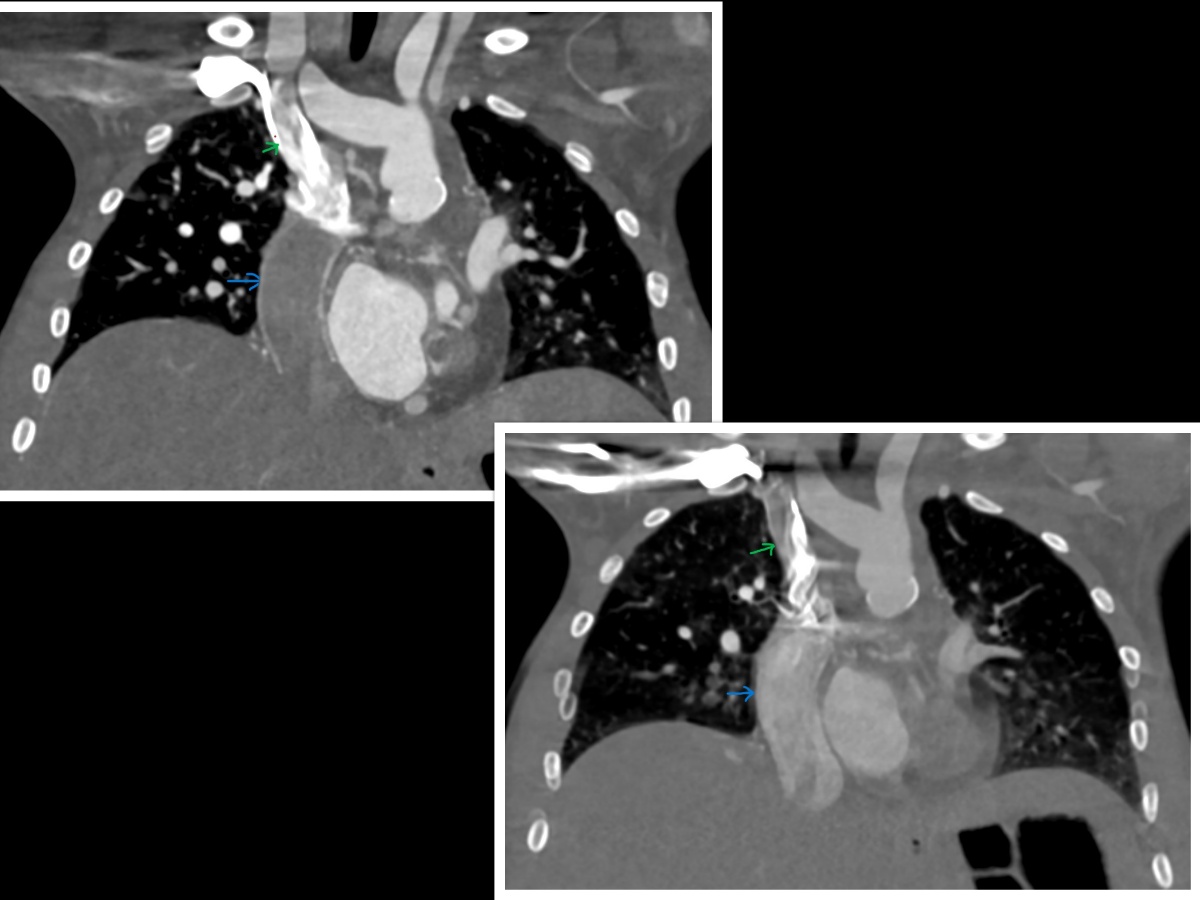
Completed Fontan on Cardiac Computed Tomography. Cardiac computed tomography of a completed Fontan palliation in a hypoplastic left heart syndrome patient. The bidirectional Glenn (green arrow) and the Fontan conduit (blue arrow) are shown.
Contributed by A Thomas, MD
(Click Video to Play)
Completed Fontan on Cardiac Magnetic Resonance Angiography. Cardiac magnetic resonance angiography in a patient status post Fontan palliation demonstrates the patent bidirectional Glenn (dashed arrow) and the Fontan conduit (solid arrow).
Contributed by A Thomas, MD
References
Yabrodi M, Mastropietro CW. Hypoplastic left heart syndrome: from comfort care to long-term survival. Pediatric research. 2017 Jan:81(1-2):142-149. doi: 10.1038/pr.2016.194. Epub 2016 Oct 4 [PubMed PMID: 27701379]
Grossfeld P, Nie S, Lin L, Wang L, Anderson RH. Hypoplastic Left Heart Syndrome: A New Paradigm for an Old Disease? Journal of cardiovascular development and disease. 2019 Feb 23:6(1):. doi: 10.3390/jcdd6010010. Epub 2019 Feb 23 [PubMed PMID: 30813450]
Gobergs R, Salputra E, Lubaua I. Hypoplastic left heart syndrome: a review. Acta medica Lituanica. 2016:23(2):86-98. doi: 10.6001/actamedica.v23i2.3325. Epub [PubMed PMID: 28356795]
Roeleveld PP, Axelrod DM, Klugman D, Jones MB, Chanani NK, Rossano JW, Costello JM. Hypoplastic left heart syndrome: from fetus to fontan. Cardiology in the young. 2018 Nov:28(11):1275-1288. doi: 10.1017/S104795111800135X. Epub 2018 Sep 18 [PubMed PMID: 30223915]
Vali P, Lakshminrusimha S. The Fetus Can Teach Us: Oxygen and the Pulmonary Vasculature. Children (Basel, Switzerland). 2017 Aug 3:4(8):. doi: 10.3390/children4080067. Epub 2017 Aug 3 [PubMed PMID: 28771211]
Green A, Pye S, Yetman AT. The physiologic basis for and nursing considerations in the use of subatmospheric concentrations of oxygen in HLHS. Advances in neonatal care : official journal of the National Association of Neonatal Nurses. 2002 Aug:2(4):177-86 [PubMed PMID: 12881932]
Level 3 (low-level) evidenceRao PS. Management of Congenital Heart Disease: State of the Art-Part II-Cyanotic Heart Defects. Children (Basel, Switzerland). 2019 Apr 4:6(4):. doi: 10.3390/children6040054. Epub 2019 Apr 4 [PubMed PMID: 30987364]
Yun SW. Congenital heart disease in the newborn requiring early intervention. Korean journal of pediatrics. 2011 May:54(5):183-91. doi: 10.3345/kjp.2011.54.5.183. Epub 2011 May 31 [PubMed PMID: 21829408]
Mackie SA, Aiyagari R, Zampi JD. Balloon atrial septostomy by a right internal jugular venous approach in a newborn with hypoplastic left heart syndrome with a restrictive atrial septum. Congenital heart disease. 2014 Sep-Oct:9(5):E140-2. doi: 10.1111/chd.12108. Epub 2013 Jun 18 [PubMed PMID: 23773545]
Level 3 (low-level) evidenceFillipps DJ, Bucciarelli RL. Cardiac evaluation of the newborn. Pediatric clinics of North America. 2015 Apr:62(2):471-89. doi: 10.1016/j.pcl.2014.11.009. Epub [PubMed PMID: 25836709]
Mawson IE, Babu PL, Simpson JM, Fox GF. Pulse oximetry findings in newborns with antenatally diagnosed congenital heart disease. European journal of pediatrics. 2018 May:177(5):683-689. doi: 10.1007/s00431-018-3093-2. Epub 2018 Feb 5 [PubMed PMID: 29404717]
Zakaria D, Tang X, Bhakta R, ElHassan NO, Prodhan P. Chromosomal Abnormalities Affect the Surgical Outcome in Infants with Hypoplastic Left Heart Syndrome: A Large Cohort Analysis. Pediatric cardiology. 2018 Jan:39(1):11-18. doi: 10.1007/s00246-017-1717-3. Epub 2017 Sep 18 [PubMed PMID: 28921168]
Nguyen T, Miller M, Gonzalez J, Nardell K, Galas J, John JB, Timofeev S, Lambou R, Douglas K, Marx G. Echocardiography of hypoplastic left heart syndrome. Cardiology in the young. 2011 Dec:21 Suppl 2():28-37. doi: 10.1017/S1047951111001557. Epub [PubMed PMID: 22152526]
Bardo DM, Frankel DG, Applegate KE, Murphy DJ, Saneto RP. Hypoplastic left heart syndrome. Radiographics : a review publication of the Radiological Society of North America, Inc. 2001 May-Jun:21(3):705-17 [PubMed PMID: 11353117]
Grosse-Wortmann L, Yun TJ, Al-Radi O, Kim S, Nii M, Lee KJ, Redington A, Yoo SJ, van Arsdell G. Borderline hypoplasia of the left ventricle in neonates: insights for decision-making from functional assessment with magnetic resonance imaging. The Journal of thoracic and cardiovascular surgery. 2008 Dec:136(6):1429-36. doi: 10.1016/j.jtcvs.2008.04.027. Epub 2008 Sep 6 [PubMed PMID: 19114185]
Banka P, Schaetzle B, Komarlu R, Emani S, Geva T, Powell AJ. Cardiovascular magnetic resonance parameters associated with early transplant-free survival in children with small left hearts following conversion from a univentricular to biventricular circulation. Journal of cardiovascular magnetic resonance : official journal of the Society for Cardiovascular Magnetic Resonance. 2014 Oct 7:16(1):73. doi: 10.1186/s12968-014-0073-1. Epub 2014 Oct 7 [PubMed PMID: 25314952]
Jones BO, Ditchfield MR, Cahoon GD, Hardy P, d'Udekem Y, Brizard CP, Penny DJ, Cheung MM. Cardiac magnetic resonance imaging prior to bidirectional cavopulmonary connection in hypoplastic left heart syndrome. Heart, lung & circulation. 2010 Sep:19(9):535-40. doi: 10.1016/j.hlc.2010.03.009. Epub 2010 May 7 [PubMed PMID: 20452284]
Brown DW, Gauvreau K, Powell AJ, Lang P, Colan SD, Del Nido PJ, Odegard KC, Geva T. Cardiac magnetic resonance versus routine cardiac catheterization before bidirectional glenn anastomosis in infants with functional single ventricle: a prospective randomized trial. Circulation. 2007 Dec 4:116(23):2718-25 [PubMed PMID: 18025538]
Level 1 (high-level) evidenceHan BK, Vezmar M, Lesser JR, Michalak G, Grant K, Dassenko D, Maresh J, Overman DM. Selective use of cardiac computed tomography angiography: an alternative diagnostic modality before second-stage single ventricle palliation. The Journal of thoracic and cardiovascular surgery. 2014 Oct:148(4):1548-54. doi: 10.1016/j.jtcvs.2014.04.047. Epub 2014 May 5 [PubMed PMID: 24930614]
Rathod RH, Prakash A, Kim YY, Germanakis IE, Powell AJ, Gauvreau K, Geva T. Cardiac magnetic resonance parameters predict transplantation-free survival in patients with fontan circulation. Circulation. Cardiovascular imaging. 2014 May:7(3):502-9. doi: 10.1161/CIRCIMAGING.113.001473. Epub 2014 Mar 11 [PubMed PMID: 24619103]
Rosti L, Giamberti A, Chessa M, Butera G, Pomè G, Braga M, Carminati M, Frigiola A. Pattern of cerebral ultrasound in neonatal heart surgery. La Pediatria medica e chirurgica : Medical and surgical pediatrics. 2011 May-Jun:33(3):124-8 [PubMed PMID: 22145295]
Akkinapally S, Hundalani SG, Kulkarni M, Fernandes CJ, Cabrera AG, Shivanna B, Pammi M. Prostaglandin E1 for maintaining ductal patency in neonates with ductal-dependent cardiac lesions. The Cochrane database of systematic reviews. 2018 Feb 27:2(2):CD011417. doi: 10.1002/14651858.CD011417.pub2. Epub 2018 Feb 27 [PubMed PMID: 29486048]
Level 1 (high-level) evidenceConnor JA, Thiagarajan R. Hypoplastic left heart syndrome. Orphanet journal of rare diseases. 2007 May 11:2():23 [PubMed PMID: 17498282]
McHugh KE, Hillman DG, Gurka MJ, Gutgesell HP. Three-stage palliation of hypoplastic left heart syndrome in the University HealthSystem Consortium. Congenital heart disease. 2010 Jan-Feb:5(1):8-15. doi: 10.1111/j.1747-0803.2009.00367.x. Epub [PubMed PMID: 20136852]
Goldberg CS, Trachtenberg F, William Gaynor J, Mahle WT, Ravishankar C, Schwartz SM, Cnota JF, Ohye RG, Gongwer R, Taylor M, Paridon S, Frommelt PC, Afton K, Atz AM, Burns KM, Detterich JA, Hill KD, Cabrera AG, Lewis AB, Pizarro C, Shah A, Sharma B, Newburger JW, Pediatric Heart Network Investigators. Longitudinal Follow-Up of Children With HLHS and Association Between Norwood Shunt Type and Long-Term Outcomes: The SVR III Study. Circulation. 2023 Oct 24:148(17):1330-1339. doi: 10.1161/CIRCULATIONAHA.123.065192. Epub 2023 Oct 5 [PubMed PMID: 37795623]
Nwankwo UT, Morell EM, Trucco SM, Morell VO, Kreutzer J. Hybrid Strategy for Neonates With Ductal-Dependent Systemic Circulation at High Risk for Norwood. The Annals of thoracic surgery. 2018 Aug:106(2):595-601. doi: 10.1016/j.athoracsur.2018.03.007. Epub 2018 Apr 6 [PubMed PMID: 29630874]
Kaplinski M, Ittenbach RF, Hunt ML, Stephan D, Natarajan SS, Ravishankar C, Giglia TM, Rychik J, Rome JJ, Mahle M, Kennedy AT, Steven JM, Fuller SM, Nicolson SC, Spray TL, Gaynor JW, Mascio CE. Decreasing Interstage Mortality After the Norwood Procedure: A 30-Year Experience. Journal of the American Heart Association. 2020 Oct 20:9(19):e016889. doi: 10.1161/JAHA.120.016889. Epub 2020 Sep 23 [PubMed PMID: 32964778]
Pizarro C, Derby CD, Baffa JM, Murdison KA, Radtke WA. Improving the outcome of high-risk neonates with hypoplastic left heart syndrome: hybrid procedure or conventional surgical palliation? European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic Surgery. 2008 Apr:33(4):613-8. doi: 10.1016/j.ejcts.2007.12.042. Epub 2008 Feb 7 [PubMed PMID: 18261915]
Level 2 (mid-level) evidenceRychik J, Atz AM, Celermajer DS, Deal BJ, Gatzoulis MA, Gewillig MH, Hsia TY, Hsu DT, Kovacs AH, McCrindle BW, Newburger JW, Pike NA, Rodefeld M, Rosenthal DN, Schumacher KR, Marino BS, Stout K, Veldtman G, Younoszai AK, d'Udekem Y, American Heart Association Council on Cardiovascular Disease in the Young and Council on Cardiovascular and Stroke Nursing. Evaluation and Management of the Child and Adult With Fontan Circulation: A Scientific Statement From the American Heart Association. Circulation. 2019 Aug 6:140(6):e234-e284. doi: 10.1161/CIR.0000000000000696. Epub 2019 Jul 1 [PubMed PMID: 31256636]
King G, Ayer J, Celermajer D, Zentner D, Justo R, Disney P, Zannino D, d'Udekem Y. Atrioventricular Valve Failure in Fontan Palliation. Journal of the American College of Cardiology. 2019 Feb 26:73(7):810-822. doi: 10.1016/j.jacc.2018.12.025. Epub [PubMed PMID: 30784675]
Haddad RN, Bonnet D, Malekzadeh-Milani S. Transcatheter closure of extracardiac Fontan conduit fenestration using new promising materials. Journal of cardiac surgery. 2021 Nov:36(11):4381-4385. doi: 10.1111/jocs.15916. Epub 2021 Aug 25 [PubMed PMID: 34432916]
Morray BH, Albers EL, Jones TK, Kemna MS, Permut LC, Law YM. Hybrid stage 1 palliation as a bridge to cardiac transplantation in patients with high-risk single ventricle physiology. Pediatric transplantation. 2018 Dec:22(8):e13307. doi: 10.1111/petr.13307. Epub 2018 Oct 19 [PubMed PMID: 30338630]
Said SM, Qureshi MY, Taggart NW, Anderson HN, O'Leary PW, Cetta F, Alrahmani L, Cofer SA, Segura LG, Pike RB, Sharpe EE, Derleth DP, Nemergut ME, Van Dorn CS, Gleich SJ, Rose CH, Collura CA, Ruano R. Innovative 2-Step Management Strategy Utilizing EXIT Procedure for a Fetus With Hypoplastic Left Heart Syndrome and Intact Atrial Septum. Mayo Clinic proceedings. 2019 Feb:94(2):356-361. doi: 10.1016/j.mayocp.2018.08.004. Epub [PubMed PMID: 30711131]
Jacobs JP, Mayer JE Jr, Pasquali SK, Hill KD, Overman DM, St Louis JD, Kumar SR, Backer CL, Tweddell JS, Dearani JA, Jacobs ML. The Society of Thoracic Surgeons Congenital Heart Surgery Database: 2019 Update on Outcomes and Quality. The Annals of thoracic surgery. 2019 Mar:107(3):691-704. doi: 10.1016/j.athoracsur.2018.12.016. Epub 2019 Jan 11 [PubMed PMID: 30641069]
Level 2 (mid-level) evidenceSinzobahamvya N, Photiadis J, Kumpikaite D, Fink C, Blaschczok HC, Brecher AM, Asfour B. Comprehensive Aristotle score: implications for the Norwood procedure. The Annals of thoracic surgery. 2006 May:81(5):1794-800 [PubMed PMID: 16631674]
Wilson WM, Valente AM, Hickey EJ, Clift P, Burchill L, Emmanuel Y, Gibson P, Greutmann M, Grewal J, Grigg LE, Gurvitz M, Hickey K, Khairy P, Mayer JE Jr, Teo E, Vonder Muhll I, Roche SL, Silversides CK, Wald RM. Outcomes of Patients With Hypoplastic Left Heart Syndrome Reaching Adulthood After Fontan Palliation: Multicenter Study. Circulation. 2018 Feb 27:137(9):978-981. doi: 10.1161/CIRCULATIONAHA.117.031282. Epub [PubMed PMID: 29483175]
Level 2 (mid-level) evidenceGoldberg CS, Mussatto K, Licht D, Wernovsky G. Neurodevelopment and quality of life for children with hypoplastic left heart syndrome: current knowns and unknowns. Cardiology in the young. 2011 Dec:21 Suppl 2(0 2):88-92. doi: 10.1017/S104795111100165X. Epub [PubMed PMID: 22152534]
Level 2 (mid-level) evidencePeyton C. Protein-Losing Enteropathy and Plastic Bronchitis After the Fontan Operation. Critical care nurse. 2018 Dec:38(6):e5-e12. doi: 10.4037/ccn2018784. Epub [PubMed PMID: 30504504]
Gordon-Walker TT, Bove K, Veldtman G. Fontan-associated liver disease: A review. Journal of cardiology. 2019 Sep:74(3):223-232. doi: 10.1016/j.jjcc.2019.02.016. Epub 2019 Mar 28 [PubMed PMID: 30928109]
Emamaullee J, Zaidi AN, Schiano T, Kahn J, Valentino PL, Hofer RE, Taner T, Wald JW, Olthoff KM, Bucuvalas J, Fischer R. Fontan-Associated Liver Disease: Screening, Management, and Transplant Considerations. Circulation. 2020 Aug 11:142(6):591-604. doi: 10.1161/CIRCULATIONAHA.120.045597. Epub 2020 Aug 10 [PubMed PMID: 32776846]
Deal BJ, Jacobs ML. Management of the failing Fontan circulation. Heart (British Cardiac Society). 2012 Jul:98(14):1098-104. doi: 10.1136/heartjnl-2011-301133. Epub [PubMed PMID: 22739639]
Fernandes SM, McElhinney DB, Khairy P, Graham DA, Landzberg MJ, Rhodes J. Serial cardiopulmonary exercise testing in patients with previous Fontan surgery. Pediatric cardiology. 2010 Feb:31(2):175-80. doi: 10.1007/s00246-009-9580-5. Epub [PubMed PMID: 19915891]
Luo S, Honjo O. Late deaths after Fontan procedure: the next frontier. Current opinion in cardiology. 2019 Mar:34(2):156-163. doi: 10.1097/HCO.0000000000000603. Epub [PubMed PMID: 30575650]
Level 3 (low-level) evidenceMoroney E, Posma E, Dennis A, d'Udekem Y, Cordina R, Zentner D. Pregnancy in a woman with a Fontan circulation: A review. Obstetric medicine. 2018 Mar:11(1):6-11. doi: 10.1177/1753495X17737680. Epub 2017 Nov 22 [PubMed PMID: 29636807]
Canobbio MM, Warnes CA, Aboulhosn J, Connolly HM, Khanna A, Koos BJ, Mital S, Rose C, Silversides C, Stout K, American Heart Association Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Functional Genomics and Translational Biology; and Council on Quality of Care and Outcomes Research. Management of Pregnancy in Patients With Complex Congenital Heart Disease: A Scientific Statement for Healthcare Professionals From the American Heart Association. Circulation. 2017 Feb 21:135(8):e50-e87. doi: 10.1161/CIR.0000000000000458. Epub 2017 Jan 12 [PubMed PMID: 28082385]
Level 2 (mid-level) evidence