Introduction
Klinefelter syndrome is a genetic condition that affects males and is characterized by the presence of 2 or more X chromosomes. The clinical phenotype was first described in 1942 by American physician Dr. Harry Klinefelter.[1] Affected individuals often present with tall stature, small testes, gynecomastia, and azoospermia. The genetic etiology—supernumerary X chromosomes, typically 47,XXY—was identified in 1959.[2] Additional X chromosomes contribute to testicular hyalinization, fibrosis, and hypofunction, leading to genital abnormalities, most commonly hypogonadism and infertility.[3]
Neurocognitive differences began to gain broader recognition during the mid-to-late 20th century.[4] Androgen replacement, along with neuropsychological and adaptive therapies, often supports effective clinical management.[5][6] However, clinical care remains inconsistent due to delayed diagnosis, lack of standardized treatment protocols, and limited access to affordable therapies.
A similar but less common condition, Jacobs syndrome, is characterized by the 47,XYY genotype. Affected individuals are phenotypically male and may present with antisocial tendencies, asthma, autism, seizures, infertility, tall stature, macrocephaly, and hypertelorism. Please see StatPearls' companion resource, "Jacobs Syndrome," for further information.[7]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The most common karyotype in Klinefelter syndrome is 47,XXY, accounting for >90% (see Image. Klinefelter Syndrome Karyotype). Mosaic karyotypes, such as 46,XY/47,XXY, and other aneuploidies, including 48,XXXY and 49,XXXXY, have also been reported. The acquisition of the extra X chromosome is random, typically resulting from meiotic nondisjunction or postzygotic nondisjunction.
Klinefelter syndrome typically arises from a de novo nondisjunction event rather than being inherited, though inherited cases have been reported. To date, 12 cases of healthy infants born to nonmosaic fathers with Klinefelter syndrome have been documented in the literature.
A 2020 case report by Ron-El et al described the retrieval of motile spermatozoa from a nonmosaic father with Klinefelter syndrome. After fertilizing 14 eggs, 3 embryos were transferred, resulting in a triplet pregnancy. Chorionic villus sampling revealed a chromosomal analysis of 46,XX, 46,XY, and 46,XXY. One fetus affected by the 46,XXY pattern was reduced during early pregnancy, leading to the birth of 1 boy and 1 girl with normal chromosomal patterns. The report emphasized the value of genetic testing.[8] The severity of the phenotype correlates with the amount of additional X chromosome material present.
Epidemiology
Klinefelter syndrome is the most common form of aneuploidy, characterized by an abnormal number of chromosomes in the affected individual's cells. The estimated prevalence is between 1 in 500 and 1 in 1000 males.[9] Diagnosis often occurs in adulthood, as many cases remain unidentified until later in life. Recognition often begins during evaluation for specific clinical features across different life stages, such as in the following:
- Prenatal testing or observation of genital abnormalities in a newborn with hypotonia
- Documentation of learning or behavioral difficulties in an adolescent
- Evaluation for tall stature, small testicular size, or incomplete puberty in an adolescent
- Evaluation for infertility (3% of men evaluated for infertility have Klinefelter syndrome) or hypogonadism in a male adult
Up to two-thirds of individuals with Klinefelter syndrome remain undiagnosed.[10] Comparative studies suggest that Klinefelter syndrome may occur more frequently with advancing parental age, environmentally derived errors in meiosis I, or a decrease in elective terminations for prenatally diagnosed cases. Underdiagnosis is likely due to the variable phenotype, with many cases presenting with only subtle features.
An estimated quarter of individuals with Klinefelter syndrome show no discernible diagnostic features based on history or examination. With the increased use of noninvasive prenatal testing, the frequency of prenatal diagnosis is expected to rise, enabling early management by pediatricians.
Fewer than 10% of cases are detected before puberty, and only 26% to 37% are identified overall, with most remaining undiagnosed.[11][12][13] The average age of diagnosis is 30 years, and the median lifespan is reduced by 5 or 6 years compared to the general male population.[14][15]
Pathophysiology
The molecular mechanisms underlying primary testicular failure and the phenotypic heterogeneity of physical and neurocognitive features in Klinefelter syndrome remain poorly characterized. Ongoing studies attempt to determine the influence of genetic polymorphisms, skewed X-inactivation, the parental origin of the extra X chromosome, and gene dosage.
Additional X chromosome material contributes to testicular hyalinization and fibrosis, leading to primary gonadal failure that often progresses through adolescence and young adulthood. The prevalence of hypogonadism in adult males with Klinefelter syndrome ranges from 65% to 85%.[16]
The condition may manifest in newborns with microphallus, hypospadias, cryptorchidism, and small testes. Later, evolving hypogonadism results in incomplete puberty and gynecomastia. In adults, long-term hypogonadism and infertility are typical clinical manifestations.
Decreased bone density is more common in men with Klinefelter syndrome compared to men without this condition. Osteoporosis occurs in 6% to 15% of individuals, whereas osteopenia affects 25% to 48% of individuals.[17] This presentation typically begins in adolescence and results from a higher rate of bone resorption combined with decreased bone formation. Although hypogonadism partially explains this occurrence, other factors, such as reduced vitamin D levels, also play a role.[18]
The additional gene dosage of the SHOX gene in the pseudoautosomal region of the X chromosome contributes to tall stature, long limbs, and a reduced upper-lower segment ratio. The pathophysiology of neuropsychological differences observed in Klinefelter syndrome remains poorly understood.
Histopathology
In Klinefelter syndrome, the seminiferous tubules exhibit hyalinization and fibrosis in the context of gonadotropin excess, leading to firm, often undersized testes. Limited studies of testicular biopsies from individuals with Klinefelter syndrome show a reduced number of germ cells across the lifespan, with a progressive decline, particularly after puberty. In adulthood, only infrequent pockets of spermatogonia are observed.[19][20]
History and Physical
Most individuals with Klinefelter syndrome present with tall stature and long limbs, reflected in a low upper-to-lower segment ratio. Mean height typically falls at the 75th percentile, whereas weight and head circumference align with the 50th percentile. In childhood, the phallus and testes may appear relatively small (<1.9 cm). During adolescence, puberty often progresses in a discordant manner, with normal phallic growth and pubic hair development. However, the testes rarely exceed a volume of 4 mL and are characteristically firm due to hyalinization and fibrosis. Testosterone concentrations typically fall within the low to low-normal range. Gynecomastia is common, and affected individuals are at an increased risk of developing male breast cancer.[21]
A wide range of intelligence quotients (IQs) has been reported. However, the mean full-scale IQ falls between 85 and 90, with an average reduction of approximately 15 points.[22] Verbal difficulties often stem from deficits in expressive language and auditory processing.[23] Behavioral features may include immaturity, insecurity, shyness, poor judgment, and attention deficit hyperactivity disorder. Individuals with Klinefelter syndrome are at a higher risk of anxiety and depression, often related to body image concerns, interpersonal challenges, and infertility.[24][25][26][27]
Between 20% and 50% of individuals with Klinefelter syndrome experience intention tremors. Approximately 10% of affected males meet the criteria for autism spectrum disorder, supporting the need for routine autism screening in this population.[28]
Around two-thirds of individuals with Klinefelter syndrome require remedial speech therapy for language deficits. Any male receiving speech and language therapy should be evaluated for Klinefelter syndrome.[29] Additional complications may involve multiple organ systems, often identified during history and physical examination. (Please refer to the Complications section for more information.)
Evaluation
The initial evaluation of Klinefelter syndrome often involves assessing for hypogonadism or infertility. Levels of the gonadotropins—follicle-stimulating hormone (FSH) and luteinizing hormone (LH)—are typically elevated in the setting of testicular hyalinization and fibrosis. However, this hormonal pattern may evolve throughout adolescence. Hypergonadotropic hypogonadism reflects primary gonadal failure.[30]
FSH levels typically exceed those of LH, although both are elevated above normal ranges. Testosterone concentrations are typically low or low-normal in adolescents and adults. Elevated sex hormone–binding globulin levels reduce free serum testosterone, even when total testosterone appears within normal limits. Most adults with Klinefelter syndrome exhibit hypogonadism, although not universally. Serum estradiol concentrations are often high-normal or elevated, and the estradiol-to-testosterone ratio remains consistently increased, contributing to gynecomastia.[31]
Inhibin B is typically undetectable, and anti-Müllerian hormone levels are typically low in adults with Klinefelter syndrome, reflecting abnormal Sertoli cell function.[32][33][34] Both biomarkers typically remain within normal ranges during childhood but become abnormal after puberty.
Noninvasive prenatal testing for cell-free fetal DNA can detect sex chromosome abnormalities. Published positive predictive values for identifying Klinefelter syndrome through noninvasive prenatal testing range from 67% to 78%.[35] Noninvasive prenatal testing analyzes small fragments of fetal DNA circulating in maternal serum.[36] Any suspected case requires additional prenatal or postnatal testing for confirmation.
A definitive diagnosis of Klinefelter syndrome typically involves prenatal or postnatal karyotype analysis or chromosomal microarray testing. Referral to a reference laboratory with expertise in these examinations is recommended. Confirmation by karyotyping requires analysis of at least 20 cultured metaphase lymphocytes.[37] Using fluorescence in situ hybridization can improve diagnostic accuracy.[38]
A baseline dual-energy X-ray absorptiometry scan is recommended due to the higher prevalence of reduced bone mineral density and increased risk of osteoporosis in affected individuals. Contributing factors include hypogonadism, vitamin D deficiency, and altered bone remodeling.
Treatment / Management
Earlier diagnosis, often prenatally, allows timely developmental evaluation and early intervention to support neuropsychological outcomes. Speech-language and motor delays affect 50% to 75% of individuals, warranting proactive screening and early therapy. Unaddressed speech delays can impair self-expression, reduce frustration tolerance, and contribute to behavioral challenges. Hypotonia, hypermobility, pes planus, and genu valgum may interfere with motor milestones, such as handwriting and self-care, requiring physical or occupational therapy and adaptive interventions, including orthotics. Common behavioral and mental health concerns include anxiety and depression, and referral to behavioral health services can provide substantial benefit.
Supervised testosterone supplementation by a pediatric endocrinologist may mitigate physical manifestations traditionally associated with this condition. Hypogonadism often begins in utero, contributing to underdeveloped genitalia, cryptorchidism, reduced germ cell populations, small testes, and an attenuated minipuberty in infancy. Some healthcare providers administer testosterone during the first few postnatal months to treat microphallus. Retrospective data suggest potential cognitive and behavioral benefits, although prospective studies are ongoing.[39][40]
Infants with cryptorchidism or an inguinal hernia should undergo appropriate surgical intervention. The American Urological Association does not recommend hormonal treatment for cryptorchidism due to its low success rate and the lack of evidence supporting long-term benefits.[41] However, some clinicians argue that hormonal therapy poses minimal risk, may enhance future fertility, and could eliminate the need for surgery in a select subset of patients.[42][43][44](B3)
All affected individuals should undergo screening for autism spectrum disorder. Additional concerns may include impaired bone mineralization, significantly delayed language skills, diabetes mellitus, hypogonadism, and infertility. Metabolic syndrome—often associated with hypogonadism and obesity—affects approximately 44% of affected individuals.
During adolescence, most individuals enter puberty spontaneously, with endogenous testosterone supporting virilization, including normal penile growth and pubic hair development. However, reduced facial and body hair may be noted. Supplemental testosterone can reduce the severity of gynecomastia, which frequently emerges during adolescence. Other management options for gynecomastia, such as aromatase inhibitors (ineffective), tamoxifen (with limited data), or surgery (invasive and prone to recurrence), offer limited or inconsistent benefits.[45]
Testosterone plays a crucial role in bone growth and skeletal health in males. Early hormonal therapy may help prevent bone loss and reduce the risk of decreased bone mineral density.[46] Individuals with osteopenia or osteoporosis tend to respond well to treatment with testosterone, calcium, and vitamin D, particularly when therapy is sustained over an extended period.[47][48] In cases of significant demineralization, bisphosphonates or inhibitors of the receptor activator of nuclear factor κB ligand are often required. Treatment outcomes in individuals with Klinefelter syndrome are generally comparable to those observed in other populations with bone loss. Please see StatPearls' companion resource, "Osteoporosis in Males," for further information.(A1)
To ensure appropriate education, rapport, and a structured plan for monitoring and treatment, boys and their parents should engage with a pediatric endocrinologist around the onset of puberty. The decision to initiate androgen replacement should be individualized and may begin with the onset of puberty or be delayed until clear signs of hypogonadism emerge, which may not occur until late adolescence or early adulthood. Guidelines for hormone replacement in males with hypogonadism are available from the Endocrine Society and the American Urological Association.[49][50] Please see StatPearls' companion resource, "Male Hypogonadism," for further information.(A1)
Advanced reproductive technology, such as microsurgical testicular sperm extraction, has enabled up to 50% of men previously deemed infertile due to Klinefelter syndrome to father biological children.[51][52] Small pockets of sperm-producing gonadal tissue can be identified, extracted, and used for fertilization through intracytoplasmic sperm injection.
Long-term health risks in individuals with Klinefelter syndrome include conditions related to insulin resistance, such as type 2 diabetes, dyslipidemia, fatty liver disease, peripheral vascular disease, and thromboembolic disease. Careful screening and proactive preventive care are strongly recommended. Bone mineral density often declines, likely due to hypogonadism, underscoring the importance of ongoing assessment of skeletal health. Some studies also report higher rates of autoimmune disorders.
There is also an elevated risk for certain malignancies, including breast cancer, extragonadal germ cell tumors, and non-Hodgkin lymphoma.[53] Although these cancers remain rare, any concerning symptoms should prompt thorough evaluation.(B2)
Differential Diagnosis
The differential diagnosis for Klinefelter syndrome includes a range of conditions with overlapping clinical features. These clinical entities include both genetic syndromes and endocrinological disorders that can mimic or coexist with the phenotype of Klinefelter syndrome.
- Acromegaly
- Adrenogenital and gonadal-secreting tumors
- Azoospermia
- Beckwith-Wiedemann syndrome
- Constitutional gigantism
- Diabetes mellitus
- Fragile X syndrome
- Hyperprolactinemia
- Hypogonadism
- Male infertility
- Marfan syndrome
- Mosaicism
- Neurofibromatosis
- Primary testicular failure
- Sanfilippo syndrome
- Simpson-Rosan-Golabi syndrome
Accurate diagnosis requires careful consideration of these differential conditions through a comprehensive clinical evaluation, genetic testing, and appropriate screening. Early identification ensures effective management and intervention, improving outcomes for individuals affected by these syndromes.
Prognosis
Although Klinefelter syndrome cannot be cured, early diagnosis and appropriate testosterone replacement therapy can alleviate many of its adverse effects and complications. Testosterone replacement supports the development of male secondary sexual characteristics, improves bone density, and enhances focus, mood, and attention. In some cases, assisted reproductive technologies can restore fertility. However, despite these interventions, individuals with Klinefelter syndrome typically have a shortened lifespan of about 5 to 6 years.
Complications
Individuals with Klinefelter syndrome are at an increased risk for several disorders. These risks span multiple organ systems, necessitating an interprofessional approach to care.[54]
Reproductive Issues
Androgen deficiency and testicular failure are central to reproductive abnormalities in Klinefelter syndrome. Common features include azoospermia, infertility, small testes, small penis size (<8 cm), low libido, testicular failure, and gynecomastia. Sparse facial and body hair is another distinguishing trait. These issues are linked to impaired spermatogenesis, and affected individuals typically experience challenges in achieving biological paternity.
Behavioral and Neurodevelopmental Effects
Behavioral issues are also prevalent in individuals with Klinefelter syndrome. Anxiety, depression, attention deficit hyperactivity disorder, relationship issues, and autism spectrum disorder are commonly reported. Lower IQ and learning disabilities are also observed in some affected individuals.
Metabolic Abnormalities
Low testosterone levels, increased visceral fat, and greater waist circumference are associated with the elevated prevalence of type 2 diabetes in individuals with Klinefelter syndrome. Increased visceral fat in the abdominal area increases the risk of diabetes mellitus.[55] Metabolic complications also include hypertension, hyperlipidemia, and low high-density lipoprotein levels. These factors often co-occur with obesity and further increase the likelihood of cardiovascular disease.
Oncologic Complications
Individuals with Klinefelter syndrome are at elevated risk for certain cancers. A hormonal etiology has been proposed as the cause of higher rates of breast and prostate cancers. The risk of germ cell tumors, non-Hodgkin lymphoma, and lung cancer is also increased.[56]
Endocrinologic Disorders
Klinefelter syndrome is also associated with hypopituitarism, which can present with symptoms such as headaches, blurred vision, and dizziness. Individuals with this condition may experience either partial or complete hypopituitarism, which can affect multiple pituitary hormones, including growth hormone and thyroid-stimulating hormone. A case report by Rehman et al in 2018 described an individual diagnosed with Klinefelter syndrome and hypogonadism who was also found to have low levels of pituitary hormones, suggesting hypopituitarism (Endocrine Abstracts. A rare case of primary hypogonadism and partial hypopituitarism in klinefelter syndrome).
Musculoskeletal Issues
Musculoskeletal abnormalities in Klinefelter syndrome are attributed to chromosomal, hormonal, and growth-related factors. The extra X chromosome affects genes involved in bone elongation, leading to taller stature and longer legs. Delayed epiphyseal closure from testosterone deficiency causes eunuchoidal proportions. Hypogonadism reduces muscle mass and bone density, whereas normal or elevated growth hormone levels contribute to taller stature. Osteopenia and osteoporosis result from testosterone deficiency, which impairs bone formation and increases resorption. Altered vitamin D metabolism and reduced insulin-like peptide 3 levels further affect bone health. Although testosterone replacement therapy and physical rehabilitation can improve muscle mass and bone density, these interventions may not fully reverse the musculoskeletal features.
Associated Autoimmune Conditions
Individuals with Klinefelter syndrome appear to have an increased susceptibility to autoimmune diseases, potentially due to the presence of an extra X chromosome and associated hormonal imbalances. Reported conditions include systemic lupus erythematosus, rheumatoid arthritis, and autoimmune thyroid disorders, such as nodular thyroid disease. Type 1 diabetes, although less frequently recognized, has also been documented in individuals with Klinefelter syndrome. Sakurai et al described a case of Klinefelter syndrome coexisting with type 1 diabetes, highlighting the importance of routine screening for autoimmune endocrinopathies in this population.[57]
Vascular and Thromboembolic Complications
Individuals with Klinefelter syndrome have an elevated risk of venous thromboembolism, including deep vein thrombosis, pulmonary embolism, and varicose veins. This risk arises from a combination of procoagulant imbalances, hormonal factors such as testosterone deficiency, and X-linked genetic influences. Thrombophilia screening and careful monitoring are recommended, especially during testosterone therapy or periods of immobilization.
Deterrence and Patient Education
Once the diagnosis of Klinefelter syndrome is confirmed, individuals and their families should receive clear education about the condition. Clinicians should emphasize that Klinefelter syndrome is the result of a random genetic event, is not inherited, and could not have been prevented. Klinefelter syndrome is a genetic condition, not a disease, and while it cannot be cured, effective treatments are available to reduce its impact. With appropriate care, many features can be managed successfully. In many cases, fertility can now be restored using assisted reproductive technologies. Please see StatPearls' companion resources, "Male Infertility," "Assisted Reproductive Technology (ART) Techniques," and "In Vitro Fertilization," for further information.
Pearls and Other Issues
The most important points to remember when evaluating and managing Klinefelter syndrome include the following:
- Early diagnosis of Klinefelter syndrome has become more accessible, enabling timely intervention and better outcomes.
- Pediatricians should consider Klinefelter syndrome in phenotypic males presenting with developmental delays or small genitalia. Evaluation is also warranted in boys diagnosed with autism spectrum disorder or those presenting with speech and language difficulties.
- Diagnosis during infancy permits optional short-term testosterone therapy before 1 year of age to promote penile growth in cases of micropenis.[58][59][60]
- Hypogonadism, infertility, and small, firm testes define the classic adult presentation. Confirmatory testing requires karyotyping.
- Initiating testosterone replacement early remains the cornerstone of treatment.
- Fertility restoration is achievable in many individuals with Klinefelter syndrome through advanced assisted reproductive technologies.
Managing Klinefelter syndrome requires coordinated interprofessional care involving pediatricians, endocrinologists, geneticists, fertility specialists, and mental health professionals. Timely collaboration across specialties ensures comprehensive treatment that addresses hormonal, developmental, reproductive, and psychosocial needs.
Enhancing Healthcare Team Outcomes
Individuals with Klinefelter syndrome benefit significantly from coordinated, interprofessional care involving endocrinologists, urologists, neurologists, psychiatrists, geneticists, internists, pediatricians, psychologists, speech therapists, and physical therapists. This collaborative approach provides patient and caregiver education on testosterone-related adverse effects, ensures proper administration, and oversees necessary follow-up testing and monitoring. Reproductive endocrinologists and urologists specializing in male infertility may also contribute to fertility planning and management.
As individuals age, careful monitoring becomes essential to address risks such as type 2 diabetes, fatty liver, breast cancer, bone mineral density loss, and dyslipidemia. Primary care providers, including nurse practitioners, play a key role in ongoing assessments due to increased susceptibility to malignancy and chronic disease.
Long-term outcomes vary depending on mental health, with a slightly shorter lifespan than the general population. Sustained, interprofessional involvement offers the best opportunity to optimize both health and quality of life.[61]
Media
(Click Image to Enlarge)
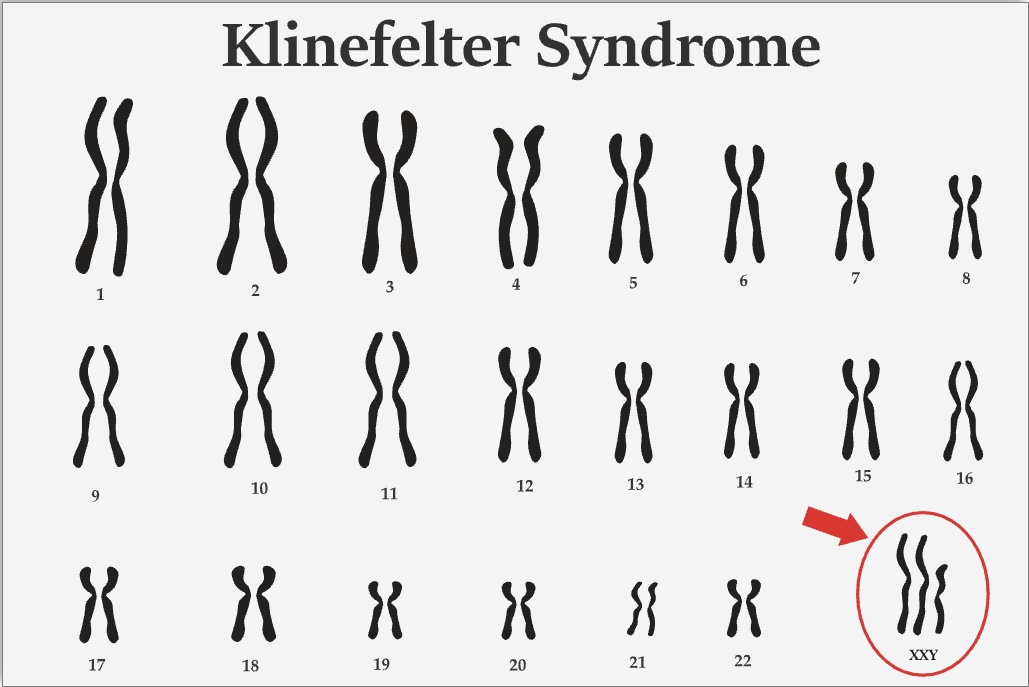
Klinefelter Syndrome Karyotype. This image shows a karyotype with an extra X chromosome (47,XXY) circled, distinguishing it from the typical 46,XY male pattern.
Shutterstock. 1455433103
References
Bonomi M, Rochira V, Pasquali D, Balercia G, Jannini EA, Ferlin A, Klinefelter ItaliaN Group (KING). Klinefelter syndrome (KS): genetics, clinical phenotype and hypogonadism. Journal of endocrinological investigation. 2017 Feb:40(2):123-134. doi: 10.1007/s40618-016-0541-6. Epub 2016 Sep 19 [PubMed PMID: 27644703]
JACOBS PA, STRONG JA. A case of human intersexuality having a possible XXY sex-determining mechanism. Nature. 1959 Jan 31:183(4657):302-3 [PubMed PMID: 13632697]
Level 3 (low-level) evidenceBelling K, Russo F, Jensen AB, Dalgaard MD, Westergaard D, Rajpert-De Meyts E, Skakkebæk NE, Juul A, Brunak S. Klinefelter syndrome comorbidities linked to increased X chromosome gene dosage and altered protein interactome activity. Human molecular genetics. 2017 Apr 1:26(7):1219-1229. doi: 10.1093/hmg/ddx014. Epub [PubMed PMID: 28369266]
Daly RF. Mental illness and patterns of behavior in 10 XXY males. The Journal of nervous and mental disease. 1969 Oct:149(4):318-27 [PubMed PMID: 5383603]
Davis S, Howell S, Wilson R, Tanda T, Ross J, Zeitler P, Tartaglia N. Advances in the Interdisciplinary Care of Children with Klinefelter Syndrome. Advances in pediatrics. 2016 Aug:63(1):15-46. doi: 10.1016/j.yapd.2016.04.020. Epub [PubMed PMID: 27426894]
Level 3 (low-level) evidenceRoss JL, Kushner H, Kowal K, Bardsley M, Davis S, Reiss AL, Tartaglia N, Roeltgen D. Androgen Treatment Effects on Motor Function, Cognition, and Behavior in Boys with Klinefelter Syndrome. The Journal of pediatrics. 2017 Jun:185():193-199.e4. doi: 10.1016/j.jpeds.2017.02.036. Epub 2017 Mar 10 [PubMed PMID: 28285751]
Sood B, Clemente Fuentes RW. Jacobs Syndrome. StatPearls. 2025 Jan:(): [PubMed PMID: 32491631]
Ron-El R, Strassburger D, Gelman-Kohan S, Friedler S, Raziel A, Appelman Z. A 47,XXY fetus conceived after ICSI of spermatozoa from a patient with non-mosaic Klinefelter's syndrome: case report. Human reproduction (Oxford, England). 2000 Aug:15(8):1804-6 [PubMed PMID: 10920107]
Level 3 (low-level) evidenceBojesen A, Juul S, Gravholt CH. Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. The Journal of clinical endocrinology and metabolism. 2003 Feb:88(2):622-6 [PubMed PMID: 12574191]
Herlihy AS, Halliday JL, Cock ML, McLachlan RI. The prevalence and diagnosis rates of Klinefelter syndrome: an Australian comparison. The Medical journal of Australia. 2011 Jan 3:194(1):24-8 [PubMed PMID: 21449864]
Akcan N, Poyrazoğlu Ş, Baş F, Bundak R, Darendeliler F. Klinefelter Syndrome in Childhood: Variability in Clinical and Molecular Findings. Journal of clinical research in pediatric endocrinology. 2018 Jun 1:10(2):100-107. doi: 10.4274/jcrpe.5121. Epub 2017 Oct 12 [PubMed PMID: 29022558]
Swerdlow AJ, Hermon C, Jacobs PA, Alberman E, Beral V, Daker M, Fordyce A, Youings S. Mortality and cancer incidence in persons with numerical sex chromosome abnormalities: a cohort study. Annals of human genetics. 2001 Mar:65(Pt 2):177-88. doi: 10.1017/S0003480001008569. Epub [PubMed PMID: 11427177]
Level 2 (mid-level) evidenceBojesen A, Kristensen K, Birkebaek NH, Fedder J, Mosekilde L, Bennett P, Laurberg P, Frystyk J, Flyvbjerg A, Christiansen JS, Gravholt CH. The metabolic syndrome is frequent in Klinefelter's syndrome and is associated with abdominal obesity and hypogonadism. Diabetes care. 2006 Jul:29(7):1591-8 [PubMed PMID: 16801584]
Level 2 (mid-level) evidenceGroth KA, Skakkebæk A, Høst C, Gravholt CH, Bojesen A. Clinical review: Klinefelter syndrome--a clinical update. The Journal of clinical endocrinology and metabolism. 2013 Jan:98(1):20-30. doi: 10.1210/jc.2012-2382. Epub 2012 Nov 1 [PubMed PMID: 23118429]
Gravholt CH, Chang S, Wallentin M, Fedder J, Moore P, Skakkebæk A. Klinefelter Syndrome: Integrating Genetics, Neuropsychology, and Endocrinology. Endocrine reviews. 2018 Aug 1:39(4):389-423. doi: 10.1210/er.2017-00212. Epub [PubMed PMID: 29438472]
Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter's syndrome. Lancet (London, England). 2004 Jul 17-23:364(9430):273-83 [PubMed PMID: 15262106]
Grande G, Graziani A, Di Mambro A, Selice R, Ferlin A. Osteoporosis and bone metabolism in patients with Klinefelter syndrome. Endocrine connections. 2023 Jul 5:12(8):. pii: e230058. doi: 10.1530/EC-23-0058. Epub 2023 Jul 5 [PubMed PMID: 37166398]
Ferlin A, Selice R, Di Mambro A, Ghezzi M, Di Nisio A, Caretta N, Foresta C. Role of vitamin D levels and vitamin D supplementation on bone mineral density in Klinefelter syndrome. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2015 Aug:26(8):2193-202. doi: 10.1007/s00198-015-3136-8. Epub 2015 May 12 [PubMed PMID: 25963234]
Corona G, Pizzocaro A, Lanfranco F, Garolla A, Pelliccione F, Vignozzi L, Ferlin A, Foresta C, Jannini EA, Maggi M, Lenzi A, Pasquali D, Francavilla S, Klinefelter ItaliaN Group (KING). Sperm recovery and ICSI outcomes in Klinefelter syndrome: a systematic review and meta-analysis. Human reproduction update. 2017 May 1:23(3):265-275. doi: 10.1093/humupd/dmx008. Epub [PubMed PMID: 28379559]
Level 1 (high-level) evidenceChehrazi M, Rahimiforoushani A, Sabbaghian M, Nourijelyani K, Sadighi Gilani MA, Hoseini M, Vesali S, Yaseri M, Alizadeh A, Mohammad K, Samani RO. Sperm Retrieval in Patients with Klinefelter Syndrome: A Skewed Regression Model Analysis. International journal of fertility & sterility. 2017 Jul-Sep:11(2):117-122. doi: 10.22074/ijfs.2017.4702. Epub 2017 Feb 16 [PubMed PMID: 28670430]
Kanakis GA, Nieschlag E. Klinefelter syndrome: more than hypogonadism. Metabolism: clinical and experimental. 2018 Sep:86():135-144. doi: 10.1016/j.metabol.2017.09.017. Epub 2018 Jan 31 [PubMed PMID: 29382506]
Linden MG, Bender BG, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics. 1995 Oct:96(4 Pt 1):672-82 [PubMed PMID: 7567329]
Level 3 (low-level) evidenceSimpson NH, Addis L, Brandler WM, Slonims V, Clark A, Watson J, Scerri TS, Hennessy ER, Bolton PF, Conti-Ramsden G, Fairfax BP, Knight JC, Stein J, Talcott JB, O'Hare A, Baird G, Paracchini S, Fisher SE, Newbury DF, SLI Consortium. Increased prevalence of sex chromosome aneuploidies in specific language impairment and dyslexia. Developmental medicine and child neurology. 2014 Apr:56(4):346-53. doi: 10.1111/dmcn.12294. Epub 2013 Oct 9 [PubMed PMID: 24117048]
Level 2 (mid-level) evidenceSrinivasan R, Wolstencroft J, Erwood M, Raymond FL, van den Bree M, Hall J, Skuse D, IMAGINE ID Consortium. Mental health and behavioural problems in children with XXYY: a comparison with intellectual disabilities. Journal of intellectual disability research : JIDR. 2019 May:63(5):477-488. doi: 10.1111/jir.12607. Epub [PubMed PMID: 30993819]
St John M, Ponchard C, van Reyk O, Mei C, Pigdon L, Amor DJ, Morgan AT. Speech and language in children with Klinefelter syndrome. Journal of communication disorders. 2019 Mar-Apr:78():84-96. doi: 10.1016/j.jcomdis.2019.02.003. Epub 2019 Feb 12 [PubMed PMID: 30822601]
van Rijn S, Swaab H. Executive dysfunction and the relation with behavioral problems in children with 47,XXY and 47,XXX. Genes, brain, and behavior. 2015 Feb:14(2):200-8. doi: 10.1111/gbb.12203. Epub [PubMed PMID: 25684214]
Bishop DV, Jacobs PA, Lachlan K, Wellesley D, Barnicoat A, Boyd PA, Fryer A, Middlemiss P, Smithson S, Metcalfe K, Shears D, Leggett V, Nation K, Scerif G. Autism, language and communication in children with sex chromosome trisomies. Archives of disease in childhood. 2011 Oct:96(10):954-9. doi: 10.1136/adc.2009.179747. Epub 2010 Jul 23 [PubMed PMID: 20656736]
Jha P, Sheth D, Ghaziuddin M. Autism spectrum disorder and Klinefelter syndrome. European child & adolescent psychiatry. 2007 Aug:16(5):305-8 [PubMed PMID: 17401614]
Level 3 (low-level) evidenceButler G, Srirangalingam U, Faithfull J, Sangster P, Senniappan S, Mitchell R. Klinefelter syndrome: going beyond the diagnosis. Archives of disease in childhood. 2023 Mar:108(3):166-171. doi: 10.1136/archdischild-2020-320831. Epub 2022 Aug 10 [PubMed PMID: 35948402]
Sizar O, Leslie SW, Schwartz J. Male Hypogonadism. StatPearls. 2025 Jan:(): [PubMed PMID: 30422528]
Maseroli E, Rastrelli G, Corona G, Boddi V, Amato AM, Mannucci E, Forti G, Maggi M. Gynecomastia in subjects with sexual dysfunction. Journal of endocrinological investigation. 2014 Jun:37(6):525-32. doi: 10.1007/s40618-014-0055-z. Epub 2014 Feb 11 [PubMed PMID: 24515298]
Level 2 (mid-level) evidenceChristiansen P, Andersson AM, Skakkebaek NE. Longitudinal studies of inhibin B levels in boys and young adults with Klinefelter syndrome. The Journal of clinical endocrinology and metabolism. 2003 Feb:88(2):888-91 [PubMed PMID: 12574229]
Anawalt BD, Bebb RA, Matsumoto AM, Groome NP, Illingworth PJ, McNeilly AS, Bremner WJ. Serum inhibin B levels reflect Sertoli cell function in normal men and men with testicular dysfunction. The Journal of clinical endocrinology and metabolism. 1996 Sep:81(9):3341-5 [PubMed PMID: 8784094]
Aksglaede L, Christiansen P, Sørensen K, Boas M, Linneberg A, Main KM, Andersson AM, Skakkebaek NE, Juul A. Serum concentrations of Anti-Müllerian Hormone (AMH) in 95 patients with Klinefelter syndrome with or without cryptorchidism. Acta paediatrica (Oslo, Norway : 1992). 2011 Jun:100(6):839-45. doi: 10.1111/j.1651-2227.2011.02148.x. Epub 2011 Feb 25 [PubMed PMID: 21251056]
Level 2 (mid-level) evidenceZhang B, Lu BY, Yu B, Zheng FX, Zhou Q, Chen YP, Zhang XQ. Noninvasive prenatal screening for fetal common sex chromosome aneuploidies from maternal blood. The Journal of international medical research. 2017 Apr:45(2):621-630. doi: 10.1177/0300060517695008. Epub 2017 Mar 30 [PubMed PMID: 28357876]
Skrzypek H, Hui L. Noninvasive prenatal testing for fetal aneuploidy and single gene disorders. Best practice & research. Clinical obstetrics & gynaecology. 2017 Jul:42():26-38. doi: 10.1016/j.bpobgyn.2017.02.007. Epub 2017 Feb 28 [PubMed PMID: 28342726]
Wiktor AE, Bender G, Van Dyke DL. Identification of sex chromosome mosaicism: is analysis of 20 metaphase cells sufficient? American journal of medical genetics. Part A. 2009 Feb:149A(2):257-9. doi: 10.1002/ajmg.a.32625. Epub [PubMed PMID: 19161142]
Abdelmoula NB, Amouri A, Portnoi MF, Saad A, Boudawara T, Mhiri MN, Bahloul A, Rebai T. Cytogenetics and fluorescence in situ hybridization assessment of sex-chromosome mosaicism in Klinefelter's syndrome. Annales de genetique. 2004 Apr-Jun:47(2):163-75 [PubMed PMID: 15183749]
Level 3 (low-level) evidenceSamango-Sprouse C, Lasutschinkow P, Powell S, Sadeghin T, Gropman A. The incidence of anxiety symptoms in boys with 47,XXY (Klinefelter syndrome) and the possible impact of timing of diagnosis and hormonal replacement therapy. American journal of medical genetics. Part A. 2019 Mar:179(3):423-428. doi: 10.1002/ajmg.a.61038. Epub 2019 Jan 13 [PubMed PMID: 30637954]
Samango-Sprouse C, Stapleton EJ, Lawson P, Mitchell F, Sadeghin T, Powell S, Gropman AL. Positive effects of early androgen therapy on the behavioral phenotype of boys with 47,XXY. American journal of medical genetics. Part C, Seminars in medical genetics. 2015 Jun:169(2):150-7. doi: 10.1002/ajmg.c.31437. Epub 2015 May 1 [PubMed PMID: 25939399]
Kolon TF, Herndon CD, Baker LA, Baskin LS, Baxter CG, Cheng EY, Diaz M, Lee PA, Seashore CJ, Tasian GE, Barthold JS, American Urological Assocation. Evaluation and treatment of cryptorchidism: AUA guideline. The Journal of urology. 2014 Aug:192(2):337-45. doi: 10.1016/j.juro.2014.05.005. Epub 2014 May 20 [PubMed PMID: 24857650]
Hadziselimovic F. Opinion: Comment on Evaluation and Treatment of Cryptorchidism: AUA/AAP and Nordic Consensus Guidelines. Urologia internationalis. 2016:96(3):249-54. doi: 10.1159/000443741. Epub 2016 Jan 30 [PubMed PMID: 26824668]
Level 3 (low-level) evidenceHadziselimović F, Huff D, Duckett J, Herzog B, Elder J, Snyder H, Buser M. Long-term effect of luteinizing hormone-releasing hormone analogue (buserelin) on cryptorchid testes. The Journal of urology. 1987 Oct:138(4 Pt 2):1043-5 [PubMed PMID: 2888905]
Radmayr C, Dogan HS, Hoebeke P, Kocvara R, Nijman R, Silay S, Stein R, Undre S, Tekgul S. Management of undescended testes: European Association of Urology/European Society for Paediatric Urology Guidelines. Journal of pediatric urology. 2016 Dec:12(6):335-343. doi: 10.1016/j.jpurol.2016.07.014. Epub 2016 Sep 15 [PubMed PMID: 27687532]
Vandeven HA, Pensler JM. Gynecomastia. StatPearls. 2025 Jan:(): [PubMed PMID: 28613563]
Ferlin A, Selice R, Carraro U, Foresta C. Testicular function and bone metabolism--beyond testosterone. Nature reviews. Endocrinology. 2013 Sep:9(9):548-54. doi: 10.1038/nrendo.2013.135. Epub 2013 Jul 16 [PubMed PMID: 23856820]
Corona G, Vena W, Pizzocaro A, Giagulli VA, Francomano D, Rastrelli G, Mazziotti G, Aversa A, Isidori AM, Pivonello R, Vignozzi L, Mannucci E, Maggi M, Ferlin A. Testosterone supplementation and bone parameters: a systematic review and meta-analysis study. Journal of endocrinological investigation. 2022 May:45(5):911-926. doi: 10.1007/s40618-021-01702-5. Epub 2022 Jan 18 [PubMed PMID: 35041193]
Level 1 (high-level) evidenceKübler A, Schulz G, Cordes U, Beyer J, Krause U. The influence of testosterone substitution on bone mineral density in patients with Klinefelter's syndrome. Experimental and clinical endocrinology. 1992:100(3):129-32 [PubMed PMID: 1305064]
Bhasin S, Brito JP, Cunningham GR, Hayes FJ, Hodis HN, Matsumoto AM, Snyder PJ, Swerdloff RS, Wu FC, Yialamas MA. Testosterone Therapy in Men With Hypogonadism: An Endocrine Society Clinical Practice Guideline. The Journal of clinical endocrinology and metabolism. 2018 May 1:103(5):1715-1744. doi: 10.1210/jc.2018-00229. Epub [PubMed PMID: 29562364]
Level 1 (high-level) evidenceMulhall JP, Trost LW, Brannigan RE, Kurtz EG, Redmon JB, Chiles KA, Lightner DJ, Miner MM, Murad MH, Nelson CJ, Platz EA, Ramanathan LV, Lewis RW. Evaluation and Management of Testosterone Deficiency: AUA Guideline. The Journal of urology. 2018 Aug:200(2):423-432. doi: 10.1016/j.juro.2018.03.115. Epub 2018 Mar 28 [PubMed PMID: 29601923]
Leslie SW, Soon-Sutton TL, Khan MAB. Male Infertility. StatPearls. 2025 Jan:(): [PubMed PMID: 32965929]
Sá R, Ferraz L, Barros A, Sousa M. The Klinefelter Syndrome and Testicular Sperm Retrieval Outcomes. Genes. 2023 Mar 4:14(3):. doi: 10.3390/genes14030647. Epub 2023 Mar 4 [PubMed PMID: 36980920]
Hasle H, Mellemgaard A, Nielsen J, Hansen J. Cancer incidence in men with Klinefelter syndrome. British journal of cancer. 1995 Feb:71(2):416-20 [PubMed PMID: 7841064]
Level 2 (mid-level) evidencedi Fraia R, Esposito D, Selvaggio LD, Allosso F, Alfano R, Rotondi M, Balercia G, Accardo G, Pasquali D. Increased prevalence of nodular thyroid disease in patients with Klinefelter syndrome. Endocrine. 2023 Sep:81(3):631-636. doi: 10.1007/s12020-023-03387-7. Epub 2023 May 6 [PubMed PMID: 37148417]
O'Connor MJ, Snyder EA, Hayes FJ. Klinefelter Syndrome and Diabetes. Current diabetes reports. 2019 Jul 31:19(9):71. doi: 10.1007/s11892-019-1197-3. Epub 2019 Jul 31 [PubMed PMID: 31367971]
Swerdlow AJ, Schoemaker MJ, Higgins CD, Wright AF, Jacobs PA, UK Clinical Cytogenetics Group. Cancer incidence and mortality in men with Klinefelter syndrome: a cohort study. Journal of the National Cancer Institute. 2005 Aug 17:97(16):1204-10 [PubMed PMID: 16106025]
Level 2 (mid-level) evidenceSakurai T, Iizuka K, Kato T, Takeda J. Type 1 Diabetes Mellitus and Klinefelter Syndrome. Internal medicine (Tokyo, Japan). 2019 Jan 15:58(2):259-262. doi: 10.2169/internalmedicine.1051-18. Epub 2018 Aug 24 [PubMed PMID: 30146555]
Wiygul J, Palmer LS. Micropenis. TheScientificWorldJournal. 2011 Jul 28:11():1462-9. doi: 10.1100/tsw.2011.135. Epub 2011 Jul 28 [PubMed PMID: 21805015]
Hatipoğlu N, Kurtoğlu S. Micropenis: etiology, diagnosis and treatment approaches. Journal of clinical research in pediatric endocrinology. 2013:5(4):217-23. doi: 10.4274/Jcrpe.1135. Epub [PubMed PMID: 24379029]
Khadilkar V, Mondkar SA. Micropenis. Indian journal of pediatrics. 2023 Jun:90(6):598-604. doi: 10.1007/s12098-023-04540-w. Epub 2023 Apr 20 [PubMed PMID: 37079255]
Thyen U, Ittermann T, Flessa S, Muehlan H, Birnbaum W, Rapp M, Marshall L, Szarras-Capnik M, Bouvattier C, Kreukels BPC, Nordenstroem A, Roehle R, Koehler B, dsd-LIFE group. Quality of health care in adolescents and adults with disorders/differences of sex development (DSD) in six European countries (dsd-LIFE). BMC health services research. 2018 Jul 5:18(1):527. doi: 10.1186/s12913-018-3342-0. Epub 2018 Jul 5 [PubMed PMID: 29976186]
Level 2 (mid-level) evidence