Introduction
Neurofibromatosis is an autosomal dominant genetic neurocutaneous disorder characterized by excessive nerve sheath tumor predisposition, most prominent in the nervous system and skin. This neurocutaneous disorder has 3 distinct types of neurofibromatosis: type 1 (NF1), type 2 (NF2), and schwannomatosis. NF1 and NF2 are the most common, while schwannomatosis is rare (see Image. Neurofibromatosis).
Neurofibromatosis Type 1 and 2
NF1, or von Recklinghausen disease, is characterized by multiple body organs with multiple (≥6) cafe-au-lait macules (CALM), neurofibromas of any type or plexiform neurofibroma, axillary or inguinal freckling, hamartomatous Lisch nodules of the iris, optic pathway glioma, and disease-specific bony dysplasia. In addition to benign and malignant tumor development, patients also have greater risks of other musculoskeletal, cardiovascular, and nervous system abnormalities. NF1 is caused by a loss-of-function mutation in 1 allele of the NF1 gene, resulting in a 50% loss of function of neurofibromin, a tumor suppressor protein ubiquitously expressed.[1] According to the National Neurofibromatosis Foundation International Database, approximately 20% of children aged 0 to 19 develop plexiform neurofibromas. Plexiform neurofibromas are benign peripheral nerve tumors with a plexiform growth pattern. They are diffuse growths involving multiple nerves and plexi with significant morbidity.[2]
NF2 has an autosomal dominant inheritance pattern and is characterized by the development of bilateral vestibular schwannomas and meningiomas. Notably, neurofibromas do not occur in this syndrome; therefore, the name NF2 is a misnomer. An international consensus has recommended the new and more accurate name of NF2-related schwannomatosis.[3] NF1 and NF2 treatment involves clinical monitoring and medical intervention when appropriate.[4][5][6]
Schwanomatosis
Schwanomatosis is the rarest of the 3 types of neurofibromatosis, with an incidence of 0.58 cases per 1,000,000 people. The presence of multiple nonintradermal peripheral and spinal schwanomas characterizes it. Localized or diffuse chronic pain or asymptomatic masses are common presentations. Vestibular schwannoma is uncommon in this entity.[7] Schwannomatosis is mostly sporadic. Approximately 15% to 25% of these patients have inherited this disorder. SMARCB1 and LZTR1 are the 2 most common gene mutations, either spontaneous or inherited with reduced penetrance. The newly revised names for these entities are SMARCB1-related schwannomatosis and LZTR1-related schwannomatosis.[3] Due to its rarity, schwanomatosis will not be elaborated on in this activity.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Neurofibromatosis Type 1
NF1 is caused by a loss-of-function mutation, either de novo or inherited, on the neurofibromin 1 (NF1) gene. Approximately half of all NF1 cases are inherited, while the other half result from de novo mutations. This gene, located on chromosome 17q11.2, encodes the protein neurofibromin. Neurofibromin is a tumor suppressor protein in the RAS/MAPK and mTOR pathways. RAS is a GDPase that activates the downstream mitogen-activated protein kinase (MAPK) and the mammalian target of rapamycin (mTOR) cell proliferation pathway.
Inadequate neurofibromin activity results in a higher risk of tumors, including malignant peripheral nerve sheath tumors (MPNST), optic pathway gliomas, and phaechromocytomas, among others. Mosaicism can occur, resulting in the segmental, generalized, or gonadal expression of the NF1 gene. The segmental NF1 gene has pigment changes, tumors, or both, and is limited to 1 or more body segments.
A diagnosis of mosaic NF1 should be considered if such lesions are present only on 1 side or in 1 body segment.[8] The generalized NF1 gene appears similar to the classic NF1 gene but lacks the typical mutation associated with the classic NF1 gene. Gonadal NF1 gene mutations occur when the mutation affects only the ova or sperm. The NF1 gene has complete penetrance with highly variable expressivity.[9]
Neurofibromatosis Type 2
A loss-of-function mutation of the NF2 gene causes NF2 and is located on chromosome 22q12, coding for the protein Merlin. Merlin is a cell membrane protein and a tumor suppressor that functions in the PI3kinase/Akt, Ras/MEK/ERK, and mTOR pathways.[10][11][12]
Epidemiology
NF1 accounts for approximately 96% of all neurofibromatosis cases. The prevalence is 1 in 3,000 births and occurs equally among both genders and races. Approximately 50% of patients have a spontaneous mutation, while the other half have an inherited mutation. A penetrance of 100% with variable expressivity is noted.
NF2 accounts for approximately 3% of all cases and has a prevalence ranging from 1 in 33,000 to 1 in 87,410 births. No gender or racial predilection has been documented. NF2 presents with variable symptoms in different families. A more severe clinical presentation is associated with a frameshift or a nonsense mutation that results in a truncated protein.[13]
Pathophysiology
Neurofibromatosis Type 1
NF1 is caused by a mutation of the NF1 gene, resulting in reduced function of its encoded protein, neurofibromin. Neurofibromin is a tumor suppressor protein involved in the downregulation of Ras signaling. The pathological hallmark of NF1 is a neurofibroma, a peripheral nerve sheath tumor that exhibits mixed nervous and fibrous components, including Schwann cells, fibroblasts, endothelial cells, mast cells, macrophages, neurons, and extracellular matrix. Cutaneous neurofibromas are present in nearly all patients with NF1, arising from dermal nerve terminals and typically developing around puberty, with their number increasing with age.
The other type of neurofibroma, plexiform neurofibroma, primarily arises from nerve plexuses in approximately 30% of patients with NF1. They grow along nerve fibers in an infiltrative fashion. Plexiform neurofibroma involves multiple nerve fascicles and has a rich vascular supply. It develops in patients younger than 5 years old and rapidly grows, resulting in significant pressure and distortion of surrounding structures. In addition to the uncontrolled proliferation of Schwann cells, NF1 also affects other cells of neurocrest origin, resulting in a variety of symptoms that affect various organ systems, including cafe-au-lait macules, axillary freckling, Lisch nodules, optic pathway glioma, and skeletal dysplasia.[14]
Neurofibromatosis Type 2
NF2 is caused by loss-of-function mutations in the NF2 gene on chromosome 22q12.2, resulting in impaired function of the encoded protein Merlin, a tumor suppressor protein. NF2 is fully penetrant. The average age of symptom onset is approximately 22. NF2 vestibular schwannoma and other tumors develop following a 2-hit hypothesis. The initial hit involves germline inactivation of an NF2 allele, followed by somatic inactivation of the NF2 gene on the opposite chromosome.[3]
Histopathology
Neurofibromatosis Type 1
Neurofibromas are benign tumors composed of mixed cell types, including Schwann cells, perineural cells, and fibroblasts. The tumors also contain mast cells, axonal processes, and a collagenous extracellular matrix. Plexiform neurofibromas are benign peripheral nerve tumors with a plexiform growth pattern. They are diffuse growths involving multiple nerves and plexi with significant morbidity due to their propensity to infiltrate surrounding structures. They also have a risk of transforming into MPNST.[2]
Neurofibromatosis Type 2
Schwannomas are the primary pathological entities associated with NF2. They most commonly arise from the eighth cranial nerve. They are benign peripheral nerve sheath tumours composed of mature Schwann cells. They consist of spindle cells with mixed Antoni A and B cellular arrangements, verocay bodies, and hyalinised vessels.[15][16]
History and Physical
Neurofibromatosis Type 1
NF1 has cutaneous and noncutaneous manifestations. CALMs are a component of the 7 diagnostic criteria for NF1. The lesions are sharply demarcated with a homogenous appearance. Axillary and groin freckling, also known as Crowe sign, is the most specific criterion for NF1.
Neurofibromas can occur anywhere on the body and can be cutaneous or internal. Dermal tumors are soft, dome-shaped tumors, but can also present as pedunculated, nodular, or plaque-like. Internal tumors are deeper and can occur around the eye, retroperitoneal, along with the gastrointestinal tract, or in the mediastinum. Additionally, neurofibromas often exhibit a buttonhole sign.
Plexiform neurofibromas are usually present from birth and are derived from the nerve sheaths (see Image. Solitary Giant Neurofibroma). They can feel like a "bag of worms." Cutaneous manifestations include scoliosis, long bone dysplasia, learning difficulties, and attention deficit hyperactivity disorder. Lisch nodules are a form of hyperpigmentation in the iris; however, they do not affect vision. Optic glioma is a tumor of the optic nerve and can affect vision, occuring in 15% of patients with neurofibromatoafe-au-latsis type 1. Patients also have generalized hyperpigmentation, blue-red, pseudoatrophic macules, juvenile xanthogranuloma, glomus tumor, melanoma, nevus anemicus, and pruritus. Patients are at increased risk for rhabdomyosarcoma, myeloid leukemia, and pheochromocytoma.
The manifestations of NF1 clinical features tend to progress with age. CALMs, bony dysplasia, and plexiform neurofibroma start during infancy. Learning deficits, ADHD, or autism spectrum disorder, and optic pathway glioma develop during early childhood. Skin freckling, Lisch nodules, and dermal and spinal neurofibromas appear and increase in number from late childhood to adolescence. MPNST and higher-grade gliomas develop in adulthood.[17]
Neurofibromatosis Type 2
NF2 patients typically present with schwannomas and meningiomas (see Image. Neurofibromatosis Type 2). Bilateral vestibular schwannoma and unilateral vestibular schwannoma occur on the superior division of the eighth cranial nerve. This is the most common type, but can occur with any cranial nerve. Involvement of the facial nerve with the vestibular schwannoma can make surgical treatment difficult. These patients typically present with tinnitus, hearing loss, and balance difficulties. Patients who have the truncated protein were found to have the disease of onset at a younger age and a higher prevalence of tumors. Younger patients typically experience symptoms at an earlier age.[18]
Evaluation
Diagnostic Criteria of Neurofibromatosis Type 1
An international consensus group revised the diagnostic criteria for NF1 in 2021, with the presence of 2 of the following being required for the diagnosis:
- Presence of ≥6 CALMs >5 mm in prepubertal individuals and >15 mm in postpubertal individuals
- Presence of ≥2 neurofibromas or ≥1 plexiform neurofibromas
- Axillary or inguinal freckling
- Optic pathway glioma
- Presence of ≥2 iris Lisch nodules or choroidal abnormalities
- Distinct bony lesions, including sphenoid dysplasia, anterolateral bowing of the tibia, or pseudoarthrosis of a long bone
- First-degree relative with NF1, or heterozygous pathogenic NF1 variant allele fraction of 50% in apparently normal tissues, eg, white blood cells [19]
Neuroimaging Manifestations of Neurofibromatosis Type 1
The following findings may be associated with NF!:
-
Nonenhancing T2/FLAIR hyperintense foci
- Commonly known as unidentified bright objects or focal areas of signal intensity (see Image. Neurofibromatosis Type I MRI).
- Locations
- Basal ganglia
- Thalami
- Mesial temporal lobes
- Internal capsule
- Splenium
- Brainstem
- Cerebellar white matter
- Pathology demonstrates areas of myelin vacuolization
- Timeline
- Typically appear around the age of 3
- Increase in number and size until approximately 12 years
- Gradually regress thereafter
-
Other lesions associated with NF1
- Optic pathway gliomas (see Image. Optic Nerve Glioma)
- Cerebral astrocytomas
- Vascular dysplasia, eg, aneurysms, moyamoya vasculopathy
- Dural ectasia
- Sphenoid wing dysplasia
-
Peripheral and cranial manifestations
- Cranial nerve schwannomas
- Peripheral neurofibromas
- Plexiform neurofibromas (see Image. Plexiform Neurofibroma)
- Malignant peripheral nerve sheath tumors
-
Spinal manifestations
- Posterior vertebral scalloping caused by dural ectasia or neurofibromas
- Scoliosis
- Lateral meningoceles
Differential diagnosis includes neurofibromatosis type 1-like syndrome, familial cafe-au-lait spots, and segmental neurofibromatosis type 1. The NF1-like syndrome was first described in 2007. Patients with an NF1-like syndrome exhibit cafe-au-lait spots, axillary freckling, and macrocephaly; however, they typically lack the NF1 genetic mutation, neurofibromas, and Lisch nodules. NF1-like syndrome is an autosomal dominant disorder resulting from a mutation in the SPRED1 gene located on chromosome 15. Familial cafe-au-lait spots are a disorder presenting only cafe-au-lait macules.
Neuroimaging Manifestations of Neurofibromatosis Type 2
A key feature of NF2 is the presence of bilateral acoustic schwannomas, also known as vestibular schwannomas (see Image. Neurofibromatosis Type 2 MRI). Other associated lesions include meningiomas, gliomas, neurofibromas, and schwannomas of different cranial nerves.
Bilateral vestibular schwannoma is pathognomic for NF2, but not all patients with this condition have bilateral schwannoma. The NIH has established the following diagnostic criteria for NF2:
- Definitive NF2: Bilateral vestibular schwannoma (see Image. Bilateral Vestibular Schwannomas) or a first-degree relative with neurofibromatosis type 2 plus unilateral vestibular schwannoma in individuals younger than 30 or any 2 of the following:
- Meningioma
- Glioma
- Schwannoma
- Juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
- Presumptive or probable NF2
- Unilateral vestibular schwannoma in patients younger than 30 and 1 of the following:
- Meningioma
- Glioma
- Schwannoma
- Juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
- Multiple meningiomas (2 or greater) plus unilateral vestibular schwannoma in patients younger than 30 years, or 1 of the following:
- Glioma
- Schwannoma
- Juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract [20]
- Unilateral vestibular schwannoma in patients younger than 30 and 1 of the following:
Treatment / Management
Neurofibromatosis Type 1
CALMs do not require treatment; similarly, neurofibromas are benign and typically do not need treatment. Surgical excision can be performed on symptomatic lesions; however, recurrence can still occur.
Plexiform neurofibromas are often disfiguring and can be difficult to remove due to their infiltrative growth pattern. Their growth may affect the airways, which require more specialized treatment, including a tracheotomy. The growth rate of plexiform neurofibromas remains relatively constant within a single individual, and its growth after adolescence is much slower. In most cases, treatment for plexiform neurofibromas aims to improve or prevent morbidity. Plexiform neurofibromas have malignant potential. An 8% to 13% risk that plexiform neurofibromas will develop into malignant peripheral nerve sheath tumors has been reported. Therefore, plexiform neurofibromas should be suspected if there is pain for more than 1 month, new neurologic deficits, a change of the neurofibroma from soft to hard, or a rapid increase in size. These malignancies are treated with wide local excision.
Imatinib, a tyrosine kinase inhibitor, has been shown to decrease the size of plexiform neurofibroma. The recent discovery of a MEK inhibitor (MEKi) targeting downstream RAS has shown promising results for patients with inoperable, symptomatic plexiform neurofibromas. Selumatinib, a MEKi treatment, results in a good partial response and a reduction in the size of the plexiform neurofibromas.[21][22]
Monitoring for any neurologic changes and referral to a neurologist is paramount. These changes can be due to tumor development. Consistent ophthalmologic evaluation is recommended for observation of the development of optic gliomas. Chemotherapy is the treatment of choice for optic gliomas. Monitoring children for difficulty learning and behavioral issues. Counseling can be beneficial for patients to provide support regarding the autosomal dominant inheritance pattern of the disease.
Neurofibromatosis Type 2
Patients with neurofibromatosis type 2 require the assessment of their hearing. Ophthalmology evaluation, magnetic resonance imaging (MRI), audiology, and brainstem-evoked potentials are essential in managing these patients. Surgery is still the first-line treatment for symptomatic tumors, but these patients have a 44% recurrence rate.
Radiation can be used, but it increases the risk of malignant transformation. Bevacizumab, a VEGF inhibitor, is a monoclonal antibody that can be used to medically treat patients with NF2. It decreased tumor size in 53% of cases and improved hearing in 57% of cases. Patients with suspected NF2 should undergo an MRI of the head and spine, with particular attention to obtaining thin cuts through the internal auditory canals. Treatment is done if the tumor is compressing the brainstem or causing hearing loss.[23][24][25]
Differential Diagnosis
The following differential diagnoses should be considered in patients with suspected NF1; therefore, genetic testing is helpful:
- Noonan syndrome (PTPN11 mutations and RAS-MARK mutations, characterized by CALM, fundal changes, and hematological malignancies)[26]
- Legius syndrome (SPRED1 mutation, characterized by café-au-lait spots, skinfold freckles, learning issues, and macrocephaly) [27]
- Constitutional mismatch repair deficiency, characterized by CALM, skin freckling, Lisch nodules, and numerous tumors or cancers [28]
- McCune-Albright syndrome
The differential diagnosis for NF2 includes nonsyndromic schwannoma and meningiomas.
Prognosis
Neurofibromatosis Type 1
The clinical features of NF1 develop and progress with age from early infancy to adulthood. The prognosis depends on the clinical features. CALMs, dermal freckling, and dermal neurofibromas affect the patient's cosmetic appearance but generally do not impact their functions. Bony dysplasia and pseudoarthrosis may affect the patient's mobility. Optic pathway gliomas often require surgical treatment, which can affect vision. Plexiform neurofibromas may cause pain, airway and spinal cord compression, dysfigurement, and, rarely, vision impairment and malignant transformation.[17]
Neurofibromatosis Type 2
Schwannomas and meningiomas are benign tumors and are generally amenable to neurosurgical therapies.
Complications
The complications associated with neurofibromatosis vary depending on the type, including:
- NF1
- Skeletal complications, including bony deformities, tibial dysplasia, and pseudoarthrosis
- Neurological problems, eg, learning disabilities, attention deficit and hyperactivity disorders (ADHD), vision loss due to optic pathway glioma, and hearing loss
- Cardiovascular conditions, eg, hypertension, congenital heart diseases, pheochromocytoma, and Moya-Moya disease
- Increased risk of multiple cancers, including MPNST, breast cancers before the age of 50, and brain tumors
- NF2: Hearing loss (primary complication)
Deterrence and Patient Education
Deterrence of neurofibromatosis is limited due to its genetic nature, but patient education plays a crucial role in early recognition, management, and family planning. Patients and families should be counseled on the autosomal dominant inheritance pattern, which carries a 50% risk of transmission to offspring. Genetic counseling is essential for affected individuals or those with a family history to make informed reproductive decisions.
Patient education should focus on recognizing early symptoms, such as café-au-lait macules, freckling, or changes in hearing, and emphasize the importance of regular monitoring for potential tumor development. Empowering patients with knowledge about their condition enhances adherence to follow-up care, improves outcomes, and supports psychosocial well-being.
Enhancing Healthcare Team Outcomes
Effective management of neurofibromatosis requires a highly coordinated interprofessional approach to deliver patient-centered care and improve long-term outcomes. Physicians and advanced practitioners, such as nurse practitioners and physician assistants, play a central role in recognizing clinical signs, initiating diagnostic evaluations, and coordinating ongoing surveillance for complications like CNS tumors, which may have a poor prognosis if not detected early. Regular follow-up by dermatologists, ophthalmologists, neurologists, and neurosurgeons is essential to monitor for neurocutaneous lesions, optic gliomas, vestibular schwannomas, and other tumor-related manifestations. Pediatricians are particularly critical in early detection and developmental monitoring of children, ensuring timely referral to specialists. Genetic counselors contribute by educating families about inheritance patterns, facilitating early diagnosis, and supporting decision-making for future pregnancies.
Nurses are integral in patient education, symptom monitoring, care navigation, and providing psychosocial support, especially as patients cope with the visible and potentially disfiguring aspects of neurofibromatosis. Pharmacists contribute to guiding safe and effective medication use, including newer therapies such as MEK inhibitors and VEGF inhibitors, as well as monitoring for adverse effects. Open and consistent communication among team members ensures timely sharing of clinical findings, promotes unified treatment planning, and reduces the risk of fragmented care. Leveraging electronic health records for shared documentation and coordinating care transitions enhances team performance and patient safety. Ultimately, a well-synchronized interprofessional strategy fosters trust, improves quality of life for patients with neurofibromatosis, and supports proactive, individualized care across the lifespan.
Media
(Click Image to Enlarge)
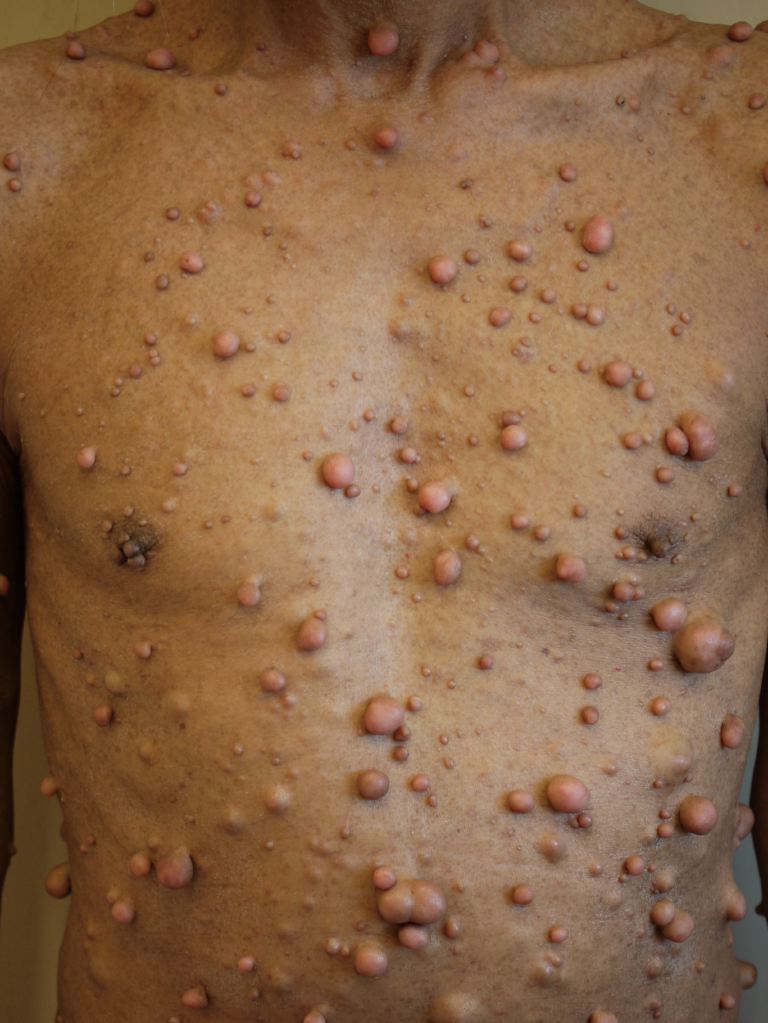
Neurofibromatosis. Neurofibromatosis encompasses three distinct disorders: neurofibromatosis type 1 (NF1), neurofibromatosis type 2 (NF2), and schwannomatosis.
(Click Image to Enlarge)

Neurofibromatosis Type 2. Neurofibromatosis type 2 patients typically present with schwannomas and meningiomas.
Contributed by S Verma, MBBS, DVD, FRCP, FAAD
(Click Image to Enlarge)
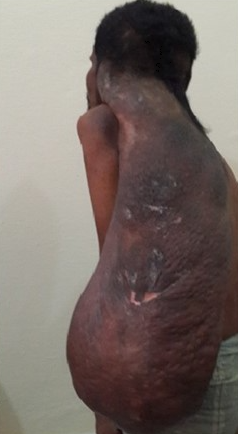
Solitary Giant Neurofibroma. Plexiform neurofibromas are usually present from birth and are derived from the nerve sheaths.
Contributed by S Munakomi, MD
(Click Image to Enlarge)
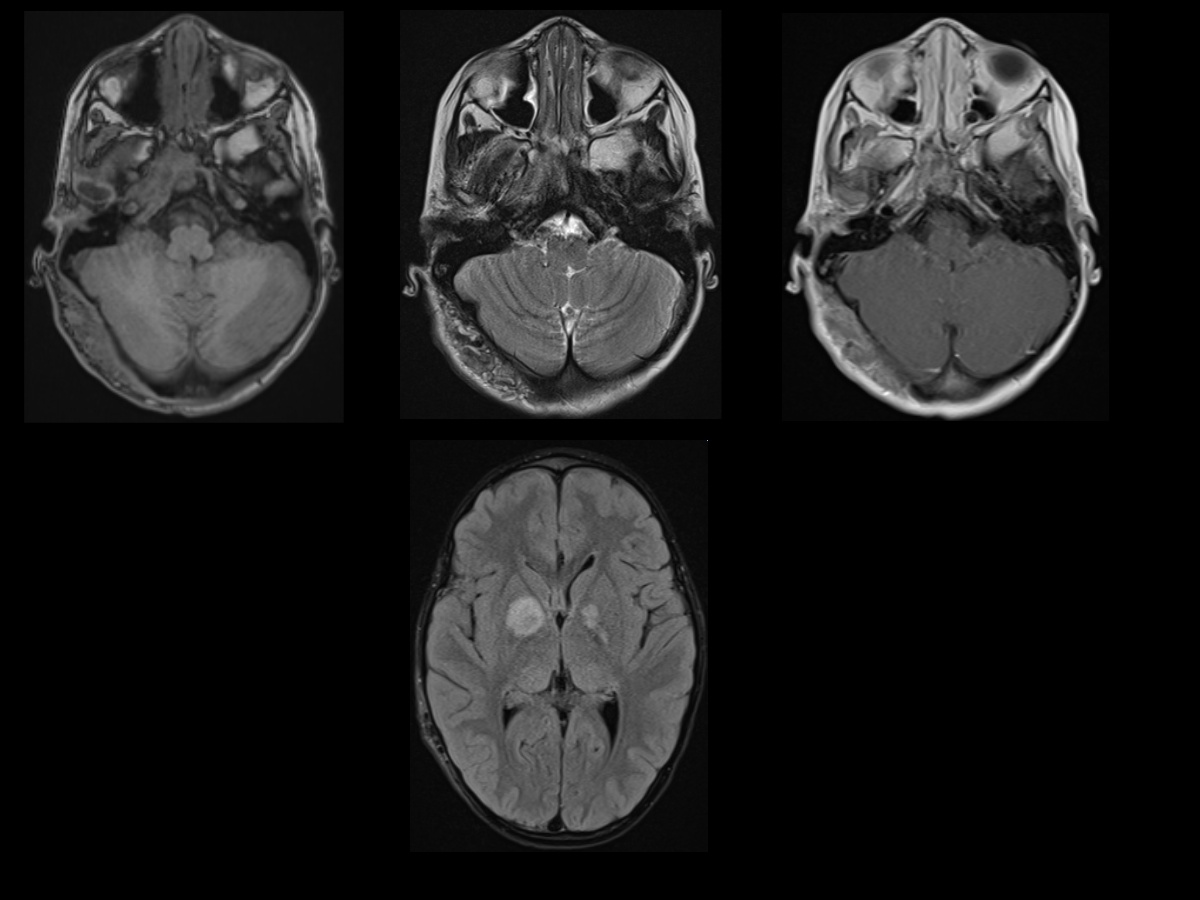
Neurofibromatosis Type I MRI. T1, T2, and postcontrast T1W MR images (top row) showing prominent soft tissue involving right occipital scalp, demonstrating heterogenous T2 signal with postcontrast enhancement, likely representing a plexiform neurofibroma. FLAIR MR image at the level of the basal ganglia (bottom row) showing areas of T2/FLAIR hyperintensities involving bilateral basal ganglia. Additional scattered areas of FLAIR hyperintensities were also seen in bilateral cerebellar hemispheres/dentate nuclei and pons (not shown), suggestive of unidentified bright objects (UBOs) or myelin vacuolization, seen in patients with a history of NF1.
Contributed by A Thomas, MD
(Click Image to Enlarge)
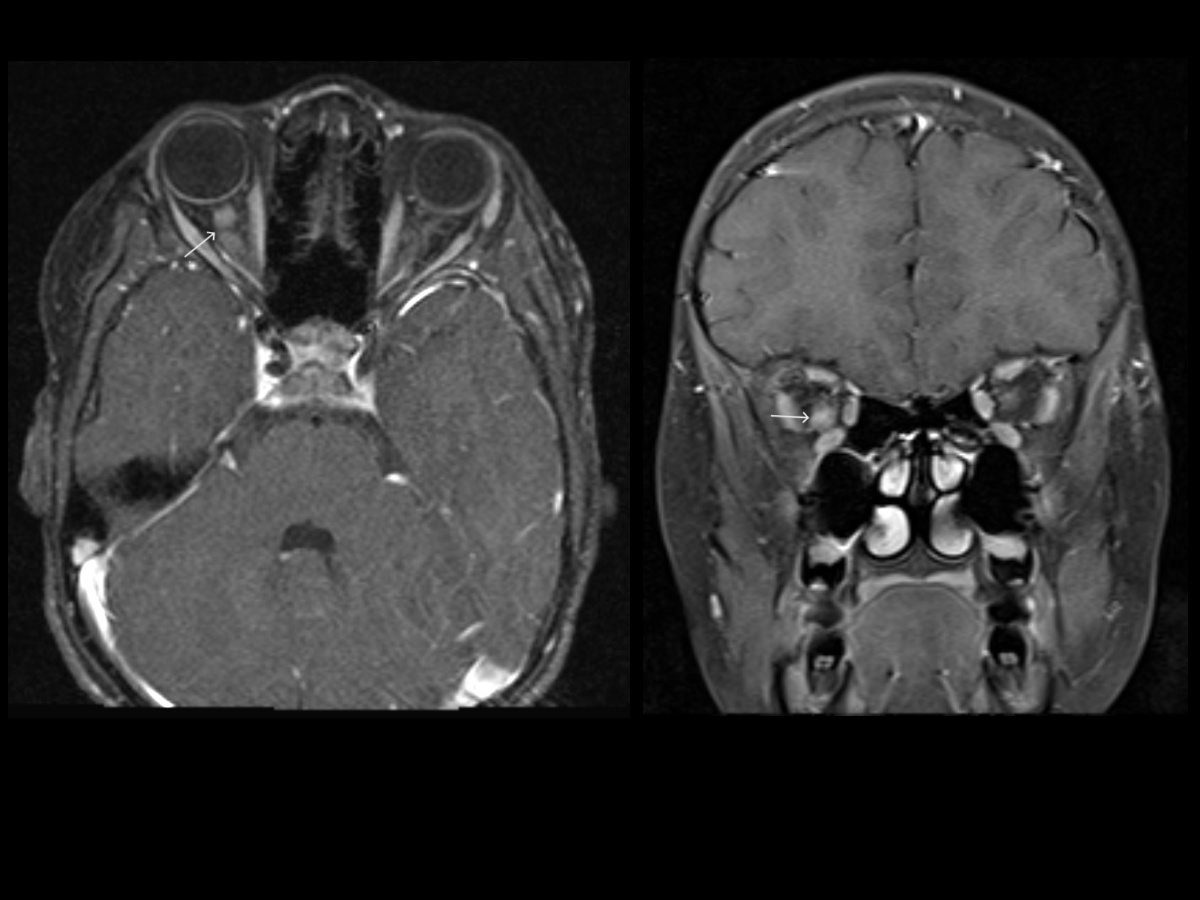
Optic Nerve Glioma. Postcontrast T1W axial and coronal MR images of the orbits demonstrating an enlarged and enhancing right optic nerve (arrows) in the intraorbital segment, consistent with right optic nerve glioma in this patient with type I neurofibromatosis.
Contributed by A Thomas, MD
(Click Image to Enlarge)
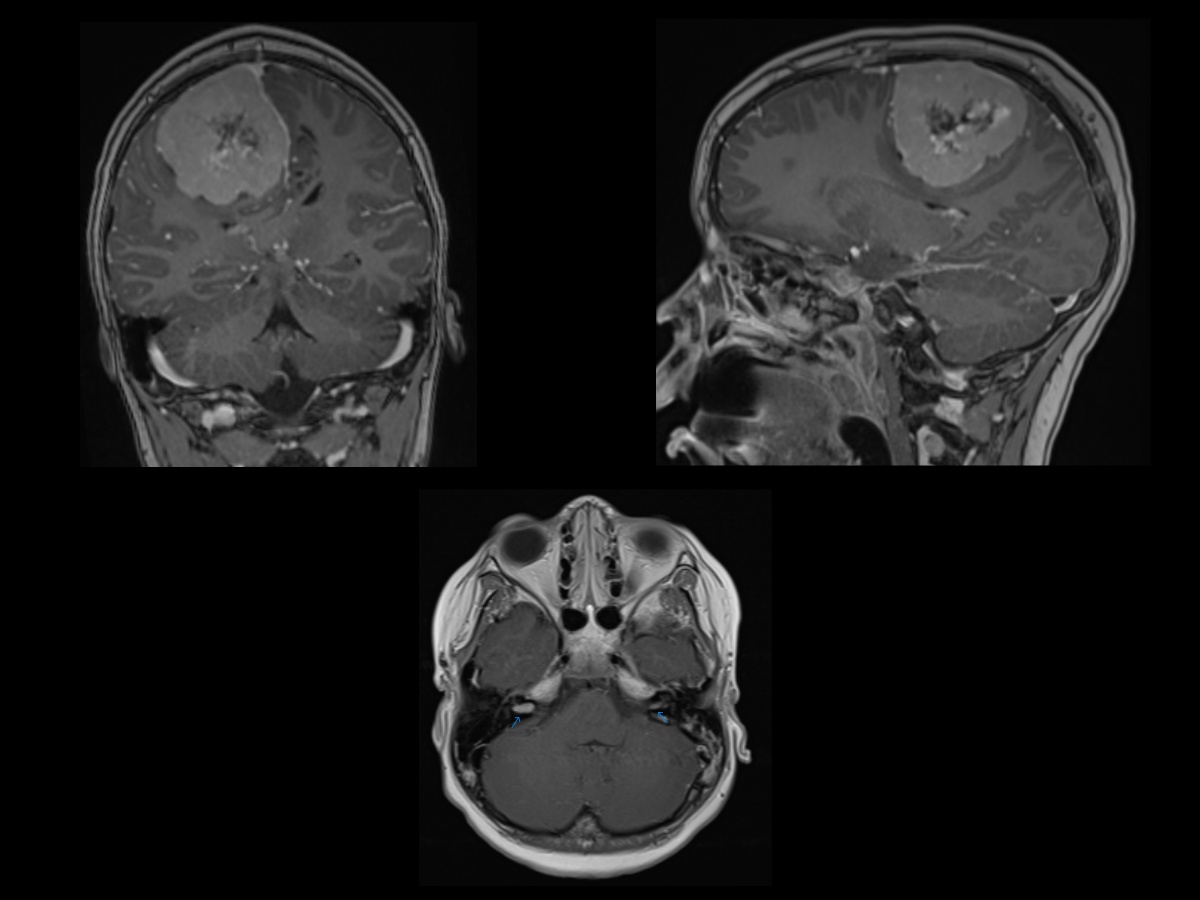
Plexiform Neurofibroma. T2W and postcontrast T1W FS axial MR images showing enhancing mass within the right cavernous sinus extending from the cavernous sinus through the superior orbital fissure into the right orbit, resulting in marked proptosis of the right globe. This likely represents a plexiform neurofibroma. Additionally, there is mild enlargement of the right temporal fossa compared to the left side in keeping with right sphenoid wing dysplasia.
Contributed by A Thomas, MD
(Click Image to Enlarge)
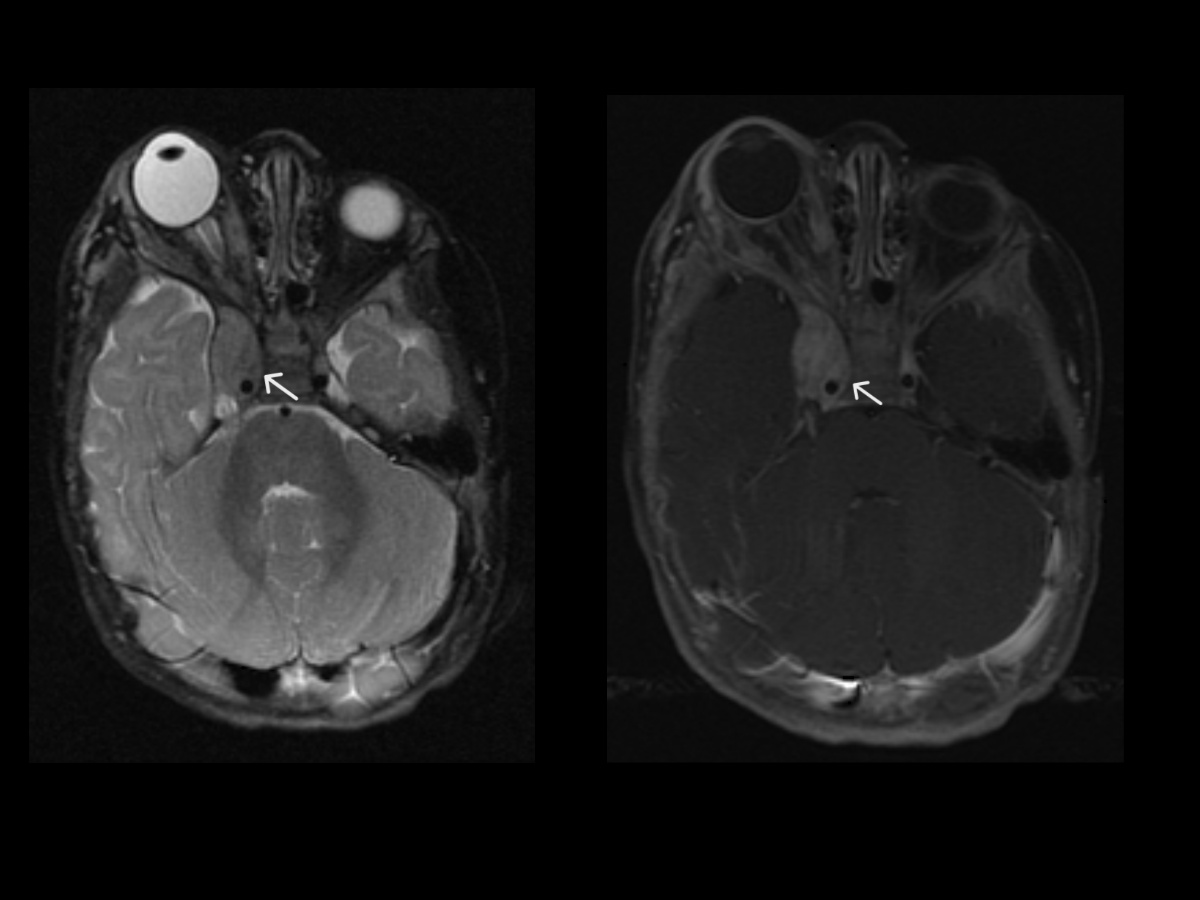
Bilateral Vestibular Schwannomas. Postcontrast T1W coronal and sagittal MR images (top row) showing a large enhancing mass at the vertex in right para-midline position with dural base on the right parietal calvarium and interhemispheric falx. Imaging features are suggestive of meningioma. Axial MR image in the same patient at the level of skull base (bottom row) showing bilateral enhancing lesions in the internal auditory canals, thin and linear on the left but bulkier on the right (arrows). Findings likely represent bilateral vestibular schwannomas and overall are essentially pathognomonic of neurofibromatosis type II.
Contributed by A Thomas, MD
(Click Image to Enlarge)
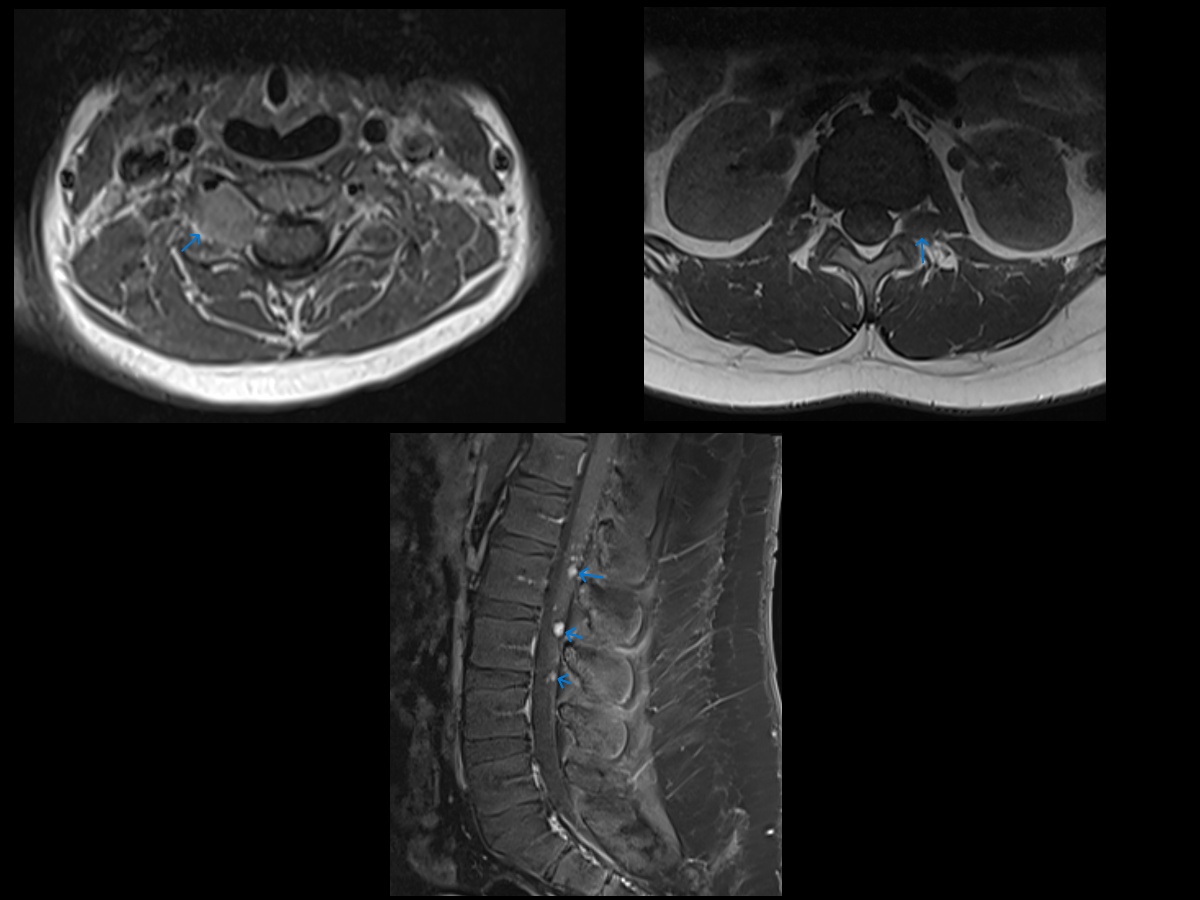
Neurofibromatosis Type 2 MRI. Axial T1W MR images at cervical and lumbar levels showing masses centered in the neural foramen. Numerous smaller masses were also seen throughout the cervical, thoracic, and lumbar levels (not shown). Postcontrast sagittal T1W FS MR image in the same patient showing numerous small enhancing nodules in the cauda equina. This patient also had a meningioma and bilateral vestibular schwannomas. This, combined with the multiple spinal masses, is essentially pathopneumonic for neurofibromatosis type II.
Contributed by A Thomas, MD
References
Poplausky D, Young JN, Tai H, Rivera-Oyola R, Gulati N, Brown RM. Dermatologic Manifestations of Neurofibromatosis Type 1 and Emerging Treatments. Cancers. 2023 May 16:15(10):. doi: 10.3390/cancers15102770. Epub 2023 May 16 [PubMed PMID: 37345107]
Copley-Merriman C, Yang X, Juniper M, Amin S, Yoo HK, Sen SS. Natural History and Disease Burden of Neurofibromatosis Type 1 with Plexiform Neurofibromas: A Systematic Literature Review. Adolescent health, medicine and therapeutics. 2021:12():55-66. doi: 10.2147/AHMT.S303456. Epub 2021 May 19 [PubMed PMID: 34040477]
Level 1 (high-level) evidenceRai P, Bathla G, Soni N, Desai A, Rao D, Vibhute P, Agarwal A. Classification of schwannomas and the new naming convention for "neurofibromatosis-2": Genetic updates and international consensus recommendation. The neuroradiology journal. 2025 Jan 9:():19714009251313510. doi: 10.1177/19714009251313510. Epub 2025 Jan 9 [PubMed PMID: 39786185]
Level 3 (low-level) evidenceBoyd KP, Korf BR, Theos A. Neurofibromatosis type 1. Journal of the American Academy of Dermatology. 2009 Jul:61(1):1-14; quiz 15-6. doi: 10.1016/j.jaad.2008.12.051. Epub [PubMed PMID: 19539839]
Ghalayani P, Saberi Z, Sardari F. Neurofibromatosis type I (von Recklinghausen's disease): A family case report and literature review. Dental research journal. 2012 Jul:9(4):483-8 [PubMed PMID: 23162593]
Level 3 (low-level) evidenceAsthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z, Lonser RR. Neurofibromatosis type 2. Lancet (London, England). 2009 Jun 6:373(9679):1974-86. doi: 10.1016/S0140-6736(09)60259-2. Epub 2009 May 22 [PubMed PMID: 19476995]
Merker VL, Esparza S, Smith MJ, Stemmer-Rachamimov A, Plotkin SR. Clinical features of schwannomatosis: a retrospective analysis of 87 patients. The oncologist. 2012:17(10):1317-22. doi: 10.1634/theoncologist.2012-0162. Epub 2012 Aug 27 [PubMed PMID: 22927469]
Level 2 (mid-level) evidenceAdam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Friedman JM. Neurofibromatosis 1. GeneReviews(®). 1993:(): [PubMed PMID: 20301288]
Wilson BN, John AM, Handler MZ, Schwartz RA. Neurofibromatosis type 1: New developments in genetics and treatment. Journal of the American Academy of Dermatology. 2021 Jun:84(6):1667-1676. doi: 10.1016/j.jaad.2020.07.105. Epub 2020 Aug 6 [PubMed PMID: 32771543]
Jouhilahti EM, Peltonen S, Heape AM, Peltonen J. The pathoetiology of neurofibromatosis 1. The American journal of pathology. 2011 May:178(5):1932-9. doi: 10.1016/j.ajpath.2010.12.056. Epub 2011 Mar 31 [PubMed PMID: 21457932]
Level 3 (low-level) evidenceEvans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet journal of rare diseases. 2009 Jun 19:4():16. doi: 10.1186/1750-1172-4-16. Epub 2009 Jun 19 [PubMed PMID: 19545378]
Cui Y, Groth S, Troutman S, Carlstedt A, Sperka T, Riecken LB, Kissil JL, Jin H, Morrison H. The NF2 tumor suppressor merlin interacts with Ras and RasGAP, which may modulate Ras signaling. Oncogene. 2019 Sep:38(36):6370-6381. doi: 10.1038/s41388-019-0883-6. Epub 2019 Jul 16 [PubMed PMID: 31312020]
Slattery WH. Neurofibromatosis type 2. Otolaryngologic clinics of North America. 2015 Jun:48(3):443-60. doi: 10.1016/j.otc.2015.02.005. Epub [PubMed PMID: 26043141]
Ge LL, Xing MY, Zhang HB, Wang ZC. Neurofibroma Development in Neurofibromatosis Type 1: Insights from Cellular Origin and Schwann Cell Lineage Development. Cancers. 2022 Sep 17:14(18):. doi: 10.3390/cancers14184513. Epub 2022 Sep 17 [PubMed PMID: 36139671]
Kresak JL, Walsh M. Neurofibromatosis: A Review of NF1, NF2, and Schwannomatosis. Journal of pediatric genetics. 2016 Jun:5(2):98-104. doi: 10.1055/s-0036-1579766. Epub 2016 Mar 9 [PubMed PMID: 27617150]
Jouhilahti EM, Peltonen S, Callens T, Jokinen E, Heape AM, Messiaen L, Peltonen J. The development of cutaneous neurofibromas. The American journal of pathology. 2011 Feb:178(2):500-5. doi: 10.1016/j.ajpath.2010.10.041. Epub [PubMed PMID: 21281783]
Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nature reviews. Disease primers. 2017 Feb 23:3():17004. doi: 10.1038/nrdp.2017.4. Epub 2017 Feb 23 [PubMed PMID: 28230061]
Ullrich NJ. Neurocutaneous Syndromes and Brain Tumors. Journal of child neurology. 2016 Oct:31(12):1399-411. doi: 10.1177/0883073815604220. Epub 2015 Oct 12 [PubMed PMID: 26459515]
Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, Blakeley J, Babovic-Vuksanovic D, Cunha KS, Ferner R, Fisher MJ, Friedman JM, Gutmann DH, Kehrer-Sawatzki H, Korf BR, Mautner VF, Peltonen S, Rauen KA, Riccardi V, Schorry E, Stemmer-Rachamimov A, Stevenson DA, Tadini G, Ullrich NJ, Viskochil D, Wimmer K, Yohay K, International Consensus Group on Neurofibromatosis Diagnostic Criteria (I-NF-DC), Huson SM, Evans DG, Plotkin SR. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genetics in medicine : official journal of the American College of Medical Genetics. 2021 Aug:23(8):1506-1513. doi: 10.1038/s41436-021-01170-5. Epub 2021 May 19 [PubMed PMID: 34012067]
Level 3 (low-level) evidenceGoutagny S, Kalamarides M. Medical treatment in neurofibromatosis type 2. Review of the literature and presentation of clinical reports. Neuro-Chirurgie. 2018 Nov:64(5):370-374. doi: 10.1016/j.neuchi.2016.09.004. Epub 2017 Feb 3 [PubMed PMID: 28162254]
Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, Weiss B, Kim A, Bornhorst M, Shah AC, Martin S, Roderick MC, Pichard DC, Carbonell A, Paul SM, Therrien J, Kapustina O, Heisey K, Clapp DW, Zhang C, Peer CJ, Figg WD, Smith M, Glod J, Blakeley JO, Steinberg SM, Venzon DJ, Doyle LA, Widemann BC. Selumetinib in Children with Inoperable Plexiform Neurofibromas. The New England journal of medicine. 2020 Apr 9:382(15):1430-1442. doi: 10.1056/NEJMoa1912735. Epub 2020 Mar 18 [PubMed PMID: 32187457]
Gross AM, Glassberg B, Wolters PL, Dombi E, Baldwin A, Fisher MJ, Kim A, Bornhorst M, Weiss BD, Blakeley JO, Whitcomb P, Paul SM, Steinberg SM, Venzon DJ, Martin S, Carbonell A, Heisey K, Therrien J, Kapustina O, Dufek A, Derdak J, Smith MA, Widemann BC. Selumetinib in children with neurofibromatosis type 1 and asymptomatic inoperable plexiform neurofibroma at risk for developing tumor-related morbidity. Neuro-oncology. 2022 Nov 2:24(11):1978-1988. doi: 10.1093/neuonc/noac109. Epub [PubMed PMID: 35467749]
Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. Journal of medical genetics. 2007 Feb:44(2):81-8 [PubMed PMID: 17105749]
Robertson KA, Nalepa G, Yang FC, Bowers DC, Ho CY, Hutchins GD, Croop JM, Vik TA, Denne SC, Parada LF, Hingtgen CM, Walsh LE, Yu M, Pradhan KR, Edwards-Brown MK, Cohen MD, Fletcher JW, Travers JB, Staser KW, Lee MW, Sherman MR, Davis CJ, Miller LC, Ingram DA, Clapp DW. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. The Lancet. Oncology. 2012 Dec:13(12):1218-24. doi: 10.1016/S1470-2045(12)70414-X. Epub 2012 Oct 23 [PubMed PMID: 23099009]
Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. The Lancet. Neurology. 2014 Aug:13(8):834-43. doi: 10.1016/S1474-4422(14)70063-8. Epub [PubMed PMID: 25030515]
Işık E, Onay H, Atik T, Solmaz AE, Özen S, Çoğulu Ö, Darcan Ş, Özkınay F. A Neurofibromatosis Noonan Syndrome Patient Presenting with Abnormal External Genitalia. Journal of clinical research in pediatric endocrinology. 2020 Mar 19:12(1):113-116. doi: 10.4274/jcrpe.galenos.2019.2019.0023. Epub 2019 May 15 [PubMed PMID: 31088041]
Denayer E, Legius E. Legius Syndrome and its Relationship with Neurofibromatosis Type 1. Acta dermato-venereologica. 2020 Mar 25:100(7):adv00093. doi: 10.2340/00015555-3429. Epub [PubMed PMID: 32147744]
Guerrini-Rousseau L, Pasmant E, Muleris M, Abbou S, Adam-De-Beaumais T, Brugieres L, Cabaret O, Colas C, Cotteret S, Decq P, Dufour C, Guillerm E, Rouleau E, Varlet P, Zili S, Vidaud D, Grill J. Neurofibromatosis type 1 mosaicism in patients with constitutional mismatch repair deficiency. Journal of medical genetics. 2024 Jan 19:61(2):158-162. doi: 10.1136/jmg-2023-109235. Epub 2024 Jan 19 [PubMed PMID: 37775264]