Introduction
Glycogen storage disease type I (GSD I), also known as Von Gierke disease, is an autosomal recessive inherited disorder that disrupts glucose production through glycogenolysis and gluconeogenesis. First documented by Dr. Edgar Von Gierke in 1929, GSD I is characterized by severe metabolic imbalances resulting from mutations that disrupt glucose-6-phosphatase (G6Pase) activity in the endoplasmic reticulum. This causes glycogen to accumulate primarily in the liver and kidneys, leading to severe fasting hypoglycemia, hyperlipidemia, lactic acidosis, hepatomegaly, and in some cases, immune dysfunction. The presentation of symptoms usually starts at around 3 to 4 months of age.[1]
GSD 1 has the following 2 main subtypes:
- GSD 1a: Deficiency of G6Pase, the enzyme that converts glucose-6-phosphate (G6P) to glucose [2]
- GSD 1b: Defect in glucose-6-phosphate translocase (G6PT), the transporter enzyme that moves G6P to the endoplasmic reticulum for hydrolysis and conversion to glucose. While G6Pase enzyme activity levels remain normal in patients with GSD 1b, the catalytic conversion rate of G6P is markedly decreased.[1][3]
Noninvasive genetic testing is the investigation of choice in patients suspected of having this disease, and treatment revolves around dietary modification. However, life expectancy remains reduced in patients with this disorder, and the risk of developing hepatic adenomas, hepatocellular carcinoma, and renal failure remains elevated. Novel therapies, eg, medications and gene therapy, are being investigated and trialed.[4][5][6]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Each disease subtype has the following genetic etiologies:
- GSD Ia results from mutations in the G6PC gene on chromosome 17q21 that encodes for G6Pase.[1][3][5] Without G6Pase activity, G6P cannot be converted into free glucose, leading to glycogen and fat accumulation in hepatocytes and renal cells.
- GSD Ib results from mutations in the SLC37A4 gene on chromosome 11q23.3 that encodes for G6PT.[1][3] The defective transport process in GSD Ib leads not only to glycogen accumulation but also to altered neutrophil function, resulting in chronic neutropenia and increased infection susceptibility.
The defect in either enzyme impairs the final step of glycogenolysis and gluconeogenesis, preventing the release of glucose from stored glycogen, triggering compensatory metabolic pathways, eg, lipolysis and proteolysis. The downstream effects include elevated free fatty acids, hyperlipidemia, hyperuricemia, and severe lactic acidosis due to the rerouting of excess G6P into glycolysis.[1][3][4][6]
Epidemiology
The incidence of GSD I in the overall population is 1 in 100,000 live births, with GSD Ia being more common (80%) than GSD 1b (20%). The Ashkenazi Jewish population has a 5 times greater prevalence compared to the rest of the population.[1][6][7] Other populations at higher risk for being carriers have been found in Mexican-Hispanics and people of Japanese decent.[8]
Carrier screening in these populations has enabled early diagnosis and proactive management, significantly improving quality of life and long-term prognosis.
Pathophysiology
The enzyme G6Pase is primarily expressed in the liver, kidney, and intestine, localized to the endoplasmic reticulum's lumen. The deficiency of G6Pase (in GSD 1a) or G6PT (in GSD 1b) leads to an accumulation of glycogen in tissues, including the liver, kidneys, and intestines. This accumulation disrupts normal cellular function and leads to hepatomegaly, nephromegaly, and metabolic derangements, eg, lactic acidosis and hyperuricemia.[1][5]
G6Pase has its active site on the luminal side of the endoplasmic reticulum. Glucose-6-phosphate translocase is responsible for translocating G6P from the cytoplasm into the endoplasmic reticulum lumen. The complex of G6Pase and G6PT catalyzes the final step of both glycogenolysis and gluconeogenesis for glucose production. Deficiency of either causes an accumulation of glycogen and fat in the liver, kidney, and intestinal mucosa.[1][3][5]
Histopathology
Although genetic testing has largely replaced the need for liver biopsy, liver histology in GSD I shows hepatocytes filled with periodic acid-Schiff (PAS)-positive and diastase-sensitive glycogen.[9] In some cases, liver biopsy may be performed in patients with unexplained hepatomegaly or when genetic testing is inconclusive.[6]
History and Physical
Some patients with GSD I may present with hypoglycemia and lactic acidosis in the neonatal period. However, they are more likely to present at 3 to 6 months of age with hepatomegaly or signs and symptoms of fasting hypoglycemia, including seizures. Thus, symptoms of hypoglycemia appear with increased intervals between feeds. Sometimes, the infant may remain asymptomatic and would present with an enlarged liver and protruding abdomen. Those left untreated would develop an appearance similar to that seen in Cushing’s syndrome, eg, short stature, round face (“doll facies”), and full cheeks.[1][3][10]
Recurrent hypoglycemia may lead to developmental delays and cognitive impairment. They have a failure to thrive along with delayed motor development. Cerebral damage resulting from recurrent hypoglycemic episodes may lead to abnormal cognitive development. In addition, patients with GSD Ib present with recurrent bacterial infections due to neutropenia.[1][3][4]
Evaluation
The diagnosis of GSD I involves a combination of clinical assessment, biochemical tests, and genetic confirmation.
Laboratory Studies
Laboratory findings typically reveal hypoglycemia, lactic acidosis, hyperuricemia, hypercholesterolemia, and hypertriglyceridemia. In GSD Ib, neutropenia of varying severity is also present.[1][3] A glucagon stimulation test should be avoided in suspected cases, as it can trigger acute acidosis and metabolic decompensation by significantly raising blood lactate levels without a corresponding increase in blood glucose.[1][3]
Genetic Studies
Genetic testing is the preferred diagnostic approach, eliminating the need for invasive liver biopsy. Full gene sequencing of G6PC (GSD Ia) and SLC37A4 (GSD Ib) detects nearly all mutations, though certain variants may be missed. In such cases, additional techniques like quantitative polymerase chain reaction (PCR), long-range PCR, multiplex ligation-dependent probe amplification, targeted array, or comparative genomic hybridization analysis can be used.[1][11]
Mutation analysis is the first-line test for confirming GSD I. If neutropenia is absent, complete G6PC sequencing is performed. In cases where liver biopsy tissue is available, G6Pase enzyme activity may also be analyzed to support the diagnosis.[1][3]
Treatment / Management
The primary management of GSD I comprises preventing acute metabolic crises, minimizing complications, ensuring normal psychological development, and improving quality of life and life expectancy.[1][4][5](A1)
Dietary Management
Management relies on a strict dietary regimen that includes:
- Frequent, high-carbohydrate meals: Regular intake of complex carbohydrates prevents hypoglycemia.[4]
- Uncooked cornstarch: A slow-releasing carbohydrate that helps maintain glucose levels during fasting, particularly overnight. Dosage is individualized and adjusted over time.[1][4]
- High-protein, low-fat diet: Supports gluconeogenesis while limiting fat intake to manage hyperlipidemia.[1]][7] (A1)
Blood glucose monitoring should be ongoing as nutritional needs change with growth. Fasting must be avoided, and small, frequent carbohydrate-rich meals with fiber are recommended, making up 60% to 70% of total calories.[1] Due to enzyme deficiencies, fructose and galactose are not metabolized to glucose-6-phosphate, necessitating a diet low in fructose and sucrose, with galactose and lactose intake limited to 1 serving per day.[7](A1)
Infants require soy-based, sugar-free formula every 2 to 3 hours to prevent hypoglycemia. As sleep duration increases beyond 3 to 4 hours, overnight hypoglycemia risk rises. Parents should be trained to use a nasogastric tube, or a G-tube may be placed for easier administration, especially during illness or feeding refusal.[6] Cornstarch remains a cornerstone treatment, providing steady glucose release. Recommended doses are 1.6 g/kg every 3 to 4 hours for young children and 1.7 to 2.5 g/kg for older children and adults. Patients should wear a medical alert bracelet, and hypoglycemia should be treated promptly with fast-acting glucose sources, including cornstarch, glucose polymers, or diabetic glucose tablets.[12]
Management of Associated Complications
In GSD Ib, neutropenia increases the risk of G-tube site infections, so granulocyte colony-stimulating factor (G-CSF) is administered before placement, with monthly CBC and spleen monitoring required.[12] Evidence now supports empagliflozin (0.3–0.4 mg/kg/d, single morning dose) for neutropenia and neutrophil dysfunction, initiated in an outpatient setting with weight-based adjustments. It should be paused during dehydration risk or major surgeries, and discontinuation of G-CSF should be attempted. If available, 1,5-AG should be monitored. Due to empagliflozin’s glycosuric effect, patients previously intolerant to starch should attempt reintroduction.[13][14][15](B3)
Safety measures, including bed-wetting detectors, infusion pump alarms, safety adapters, connectors, and secure taping, help prevent pump failures and tubing issues. Children on lactose- and sucrose-restricted diets should be assessed for nutritional deficiencies and supplemented with micronutrients.[12] Persistent lactic acidosis is treated with oral citrate or bicarbonate to alkalinize urine and reduce the risk of urolithiasis. Allopurinol helps prevent gout by lowering uric acid levels, while colchicine is preferred during acute attacks [1]. Hyperlipidemia responds partially to statins, niacin, fibrates, and fish oil, though liver transplantation has led to resolution in some cases.[9][16](A1)
Monitoring Recommendations
Routine monitoring includes blood pressure checks at every visit from infancy, with serum creatinine measured every 3 to 6 months. Persistent microalbuminuria warrants ACE inhibitor therapy, while echocardiography every 3 years is advised for pulmonary hypertension screening.[10][12] Hepatomegaly is universal due to fat and glycogen deposition, with common liver lesions including fatty infiltration, focal nodular hyperplasia, peliosis hepatis, hepatocellular adenoma (HCA), and hepatocellular carcinoma (HCC). Liver function tests should be performed every 6 to 12 months, and liver transplantation is considered for progressive, nonresponsive lesions. Females with GSD I should avoid combined oral contraceptives due to the HCA risk.[1][3][12](A1)
The European collaborative guidelines [10] recommend the following biomedical targets for GSD I management:(A1)
- Preprandial blood glucose >3.5 to 4.0 mmol/L (63–72 mg/dL)
- Urine lactate/creatinine ratio <0.06 mmol/mmol
- Serum uric acid in the high-normal range for age
- Venous blood base excess >-5 mmol/L and bicarbonate >20 mmol/L (20 meq/L)
- Serum triglycerides <6 mmol/L (531 mg/dL)
- Normal fecal alpha-1 antitrypsin for GSD Ib
- Body mass index between 0.0 and +2.0 standard deviations
Differential Diagnosis
Differentiating GSD I from other diseases that present with hepatomegaly or hypoglycemia is essential, including:
- GSD 0 (glycogen synthase deficiency)
- GSD III (glycogen debranching enzyme deficiency)
- GSD IV (branching enzyme deficiency)
- GSD VI (hepatic phosphorylase deficiency)
- GSD IX (hepatic form of phosphorylase kinase deficiency)
- GSD XI (Fanconi-Bickel syndrome due to glucose transporter protein 2 deficiency) [1][3][12][17]
- Disorders of Gluconeogenesis (Fructose-1,6-bisphosphatase deficiency)
- Primary liver disease (Hepatitis)
- Niemann-Pick B disease [18]
- Gaucher disease [18]
- Hereditary fructose intolerance
Pertinent Studies and Ongoing Trials
Dietary therapy remains the first-line treatment for GSD I. However, long-term complications such as hepatocellular adenoma (HCA), hepatocellular carcinoma (HCC), and renal failure highlight the need for alternative therapeutic approaches. Gene therapy in animal models has shown promising results, paving the way for future human trials.[5]
Gene therapy offers the potential to correct the underlying genetic defect in GSD I rather than just managing symptoms. Current research focuses on delivering functional copies of G6PC and SLC37A4 genes to liver cells to restore normal glucose metabolism and prevent glycogen accumulation. This represents a significant shift in the approach to metabolic disease treatment, with gene therapy at the forefront of potentially curative interventions.[19]
Ongoing clinical trials are evaluating the safety and efficacy of gene therapy in GSD I patients, while additional experimental approaches, such as mRNA therapy and CRISPR-Cas9 gene editing, are also being explored. These novel therapies remain investigational but hold promise for future treatment strategies.[19][20]
Dietary therapy maintains the patient's blood glucose levels and reduces the early symptoms. However, to avoid long-term complications such as HCA, HCC, and renal failure, gene therapies in GSD I mice models showed promise. In early 2018, the FDA approved the first gene therapy clinical trial at Connecticut Children’s Medical Center and UConn Health.[GeneTherapyTrial]
Prognosis
The prognosis for GSD I patients has dramatically improved with enhanced metabolic management and early diagnosis. With optimal treatment, many patients reach adulthood and lead relatively normal lives. However, they continue to face risks of liver complications, including hepatocellular adenomas and HCC, which necessitate regular imaging, such as ultrasound or MRI, for early detection [3], as well as renal complications and cardiovascular issues later in life. Liver transplantation may be considered for patients with severe liver involvement or complications that do not respond to conventional management.[12]
Complications
With early diagnosis and diligent management, the life expectancy of patients with GSD I has significantly improved. Long-term complications of untreated glycogen storage disease type I (GSD I) affect multiple organ systems, including:
- Growth and development: Poor growth and short stature are common but can improve with strict dietary management. Delayed puberty can be normalized with metabolic control.[11]
- Bone health: Osteoporosis and fractures occur due to low vitamin D and calcium intake, with metabolic control improving bone mineral density.[11] Therefore, routine monitoring of vitamin D levels, along with dual-energy x-ray absorptiometry (DXA) scans, are recommended to assess bone density and determine the need for vitamin D supplementation.[12]
- Renal complications: Proteinuria, hypertension, nephrocalcinosis, and kidney dysfunction can progress to end-stage kidney disease, requiring transplantation. Poorly controlled blood lactate, serum lipids, and uric acid levels may further increase the risk of nephropathy. Early metabolic control and angiotensin-converting enzyme (ACE) inhibitors help prevent deterioration.[11] Annual kidney ultrasounds are recommended starting in the second decade of life.[12]
- Metabolic and cardiovascular risks: Hyperuricemia can lead to gout, while systemic hypertension often appears later in life, particularly with kidney disease progression. Pulmonary hypertension may occur, especially in those with coexisting conditions. Cardiovascular disease risk remains controversial, but poor metabolic control increases the likelihood of premature atherosclerosis.[11]
- Liver: Hepatic adenomas, with a risk of malignant transformation into hepatocellular carcinoma, are common.[11] Diagnosis is often challenging due to the presence of multiple adenomas, making biopsy difficult, as well as the frequent absence of elevated tumor markers such as alpha-fetoprotein and carcinoembryonic antigen.[10] To facilitate early detection of hepatic adenomas, liver ultrasound is recommended every 12 to 24 months until age 16, followed by computed tomography or magnetic resonance imaging (MRI) every 6 to 12 months thereafter. If an adenoma is identified, follow-up imaging with ultrasound or MRI should be performed every 3 to 6 months. Given the increased risk of hepatic adenomas, female patients with GSD I should avoid taking combined oral contraceptives.[12]
- Pancreatic issues: Pancreatitis can develop due to severe hypertriglyceridemia.[11]
- Neurological and cognitive effects: Brain MRI abnormalities, cognitive impairment, and increased risk of cerebral arteriopathy have been reported, often correlating with poor metabolic control.[11]
- Hematologic disorders: Anemia is common due to dietary restrictions, chronic illness, and kidney disease. Bleeding diathesis arises from platelet dysfunction and von Willebrand factor abnormalities.[11]
- Immune dysfunction: GSD Ib is linked to neutropenia, recurrent infections, and inflammatory bowel disease. Recent evidence suggests empagliflozin may improve neutrophil function.[11]
- Reproductive and endocrine effects: Polycystic ovaries, irregular menstrual cycles, and increased thyroid autoimmunity are noted, particularly in GSD Ib.[11]
Deterrence and Patient Education
Genetic counseling for parents is highly recommended as each child of an affected carrier couple has a 25% chance of being affected and a 50% chance of being an asymptomatic carrier.[1][3]
Pearls and Other Issues
Key factors to bear in mind in the management of GSD I include:
- Early intervention with dietary management is crucial for preventing irreversible cognitive and physical damage.
- The use of G-CSF in GSD Ib is critical for reducing infection risk due to neutropenia.
- Monitoring for renal and hepatic complications should be a lifelong process.
Enhancing Healthcare Team Outcomes
Effective management of GSD I requires a comprehensive interprofessional approach, integrating the expertise of physicians, advanced practitioners, nurses, pharmacists, dietitians, genetic counselors, and specialists in hepatology and nephrology. Physicians and advanced practitioners play a key role in diagnosing GSD I, coordinating genetic testing, and overseeing long-term care plans. Nurses provide essential patient education, monitor for complications, and assist with feeding regimens, including the administration of nasogastric or gastrostomy tube feedings when needed. Pharmacists contribute by ensuring appropriate medication management, particularly for patients requiring granulocyte colony-stimulating factor (G-CSF) for neutropenia or adjunctive therapies to address metabolic imbalances. Dietitians are integral in formulating individualized nutritional plans to maintain blood glucose levels, prevent metabolic crises, and optimize growth and development, while genetic counselors support families in understanding the hereditary aspects of the disease and guide reproductive decision-making.
Interprofessional communication and coordinated care strategies are essential for improving patient safety, treatment adherence, and long-term outcomes. Regular team meetings and shared decision-making enhance the effectiveness of dietary interventions and emerging therapies, including gene therapy trials, which have shown promise in GSD I animal models and early human studies. By fostering collaboration between healthcare professionals, early intervention strategies can be implemented to prevent complications such as hepatic adenomas (HCA), hepatocellular carcinoma (HCC), and renal failure. Ensuring that families receive comprehensive, coordinated care improves patient-centered outcomes and enhances overall team performance in managing this complex disorder.
Media
(Click Image to Enlarge)
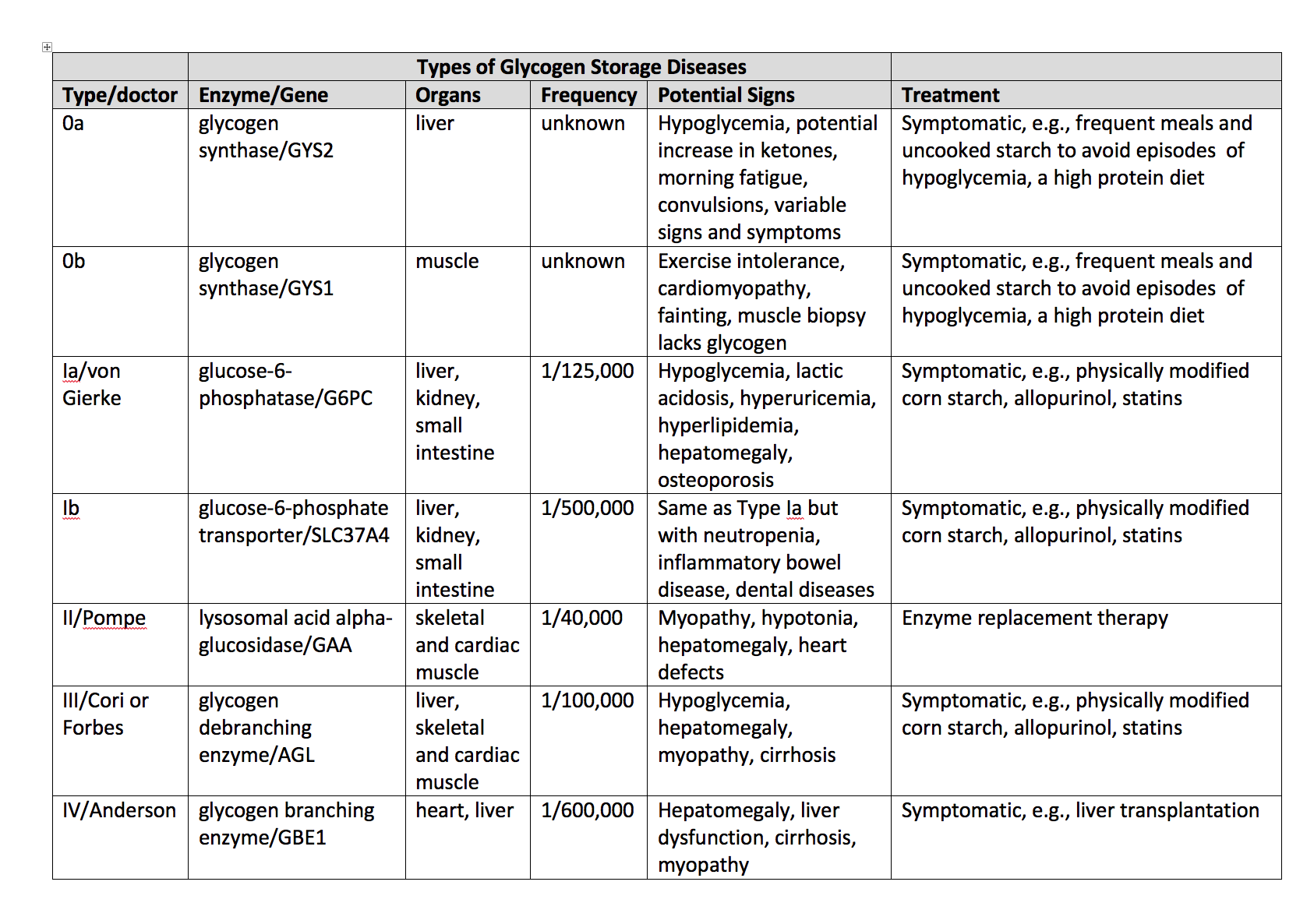
Glycogen Storage Disease Types. Other glycogen storage disorders are mainly differentiated by a lack of hypoglycemia in glycogen storage disease type II.
Contributed by WL Stone, MD
References
Kishnani PS, Austin SL, Abdenur JE, Arn P, Bali DS, Boney A, Chung WK, Dagli AI, Dale D, Koeberl D, Somers MJ, Wechsler SB, Weinstein DA, Wolfsdorf JI, Watson MS, American College of Medical Genetics and Genomics. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genetics in medicine : official journal of the American College of Medical Genetics. 2014 Nov:16(11):e1 [PubMed PMID: 25356975]
Level 1 (high-level) evidenceShieh JJ, Terzioglu M, Hiraiwa H, Marsh J, Pan CJ, Chen LY, Chou JY. The molecular basis of glycogen storage disease type 1a: structure and function analysis of mutations in glucose-6-phosphatase. The Journal of biological chemistry. 2002 Feb 15:277(7):5047-53 [PubMed PMID: 11739393]
Kishnani PS, Goldstein J, Austin SL, Arn P, Bachrach B, Bali DS, Chung WK, El-Gharbawy A, Brown LM, Kahler S, Pendyal S, Ross KM, Tsilianidis L, Weinstein DA, Watson MS, ACMG Work Group on Diagnosis and Management of Glycogen Storage Diseases Type VI and IX. Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genetics in medicine : official journal of the American College of Medical Genetics. 2019 Apr:21(4):772-789. doi: 10.1038/s41436-018-0364-2. Epub 2019 Jan 19 [PubMed PMID: 30659246]
Raza M, Arif F, Giyanwani PR, Azizullah S, Kumari S. Dietary Therapy for Von Gierke's Disease: A Case Report. Cureus. 2017 Aug 8:9(8):e1548. doi: 10.7759/cureus.1548. Epub 2017 Aug 8 [PubMed PMID: 29018645]
Level 3 (low-level) evidenceChou JY, Kim GY, Cho JH. Recent development and gene therapy for glycogen storage disease type Ia. Liver research (Beijing, China). 2017 Sep:1(3):174-180. doi: 10.1016/j.livres.2017.12.001. Epub [PubMed PMID: 29576889]
McAdams AJ, Hug G, Bove KE. Glycogen storage disease, types I to X: criteria for morphologic diagnosis. Human pathology. 1974 Jul:5(4):463-87 [PubMed PMID: 4525190]
Goldberg T, Slonim AE. Nutrition therapy for hepatic glycogen storage diseases. Journal of the American Dietetic Association. 1993 Dec:93(12):1423-30 [PubMed PMID: 8245377]
Derks TGJ, Rodriguez-Buritica DF, Ahmad A, de Boer F, Couce ML, Grünert SC, Labrune P, López Maldonado N, Fischinger Moura de Souza C, Riba-Wolman R, Rossi A, Saavedra H, Gupta RN, Valayannopoulos V, Mitchell J. Glycogen Storage Disease Type Ia: Current Management Options, Burden and Unmet Needs. Nutrients. 2021 Oct 27:13(11):. doi: 10.3390/nu13113828. Epub 2021 Oct 27 [PubMed PMID: 34836082]
Nagasaka H, Hirano K, Ohtake A, Miida T, Takatani T, Murayama K, Yorifuji T, Kobayashi K, Kanazawa M, Ogawa A, Takayanagi M. Improvements of hypertriglyceridemia and hyperlacticemia in Japanese children with glycogen storage disease type Ia by medium-chain triglyceride milk. European journal of pediatrics. 2007 Oct:166(10):1009-16 [PubMed PMID: 17206455]
Level 3 (low-level) evidenceRake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP, European Study on Glycogen Storage Disease Type I (ESGSD I). Guidelines for management of glycogen storage disease type I - European Study on Glycogen Storage Disease Type I (ESGSD I). European journal of pediatrics. 2002 Oct:161 Suppl 1():S112-9 [PubMed PMID: 12373584]
Level 1 (high-level) evidenceAdam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Bali DS, El-Gharbawy A, Austin S, Pendyal S, Kishnani PS. Glycogen Storage Disease Type I. GeneReviews(®). 1993:(): [PubMed PMID: 20301489]
Parikh NS, Ahlawat R. Glycogen Storage Disease Type I. StatPearls. 2025 Jan:(): [PubMed PMID: 30480935]
Grünert SC, Derks TGJ, Mundy H, Dalton RN, Donadieu J, Hofbauer P, Jones N, Uçar SK, LaFreniere J, Contreras EL, Pendyal S, Rossi A, Schneider B, Spiegel R, Stepien KM, Wesol-Kucharska D, Veiga-da-Cunha M, Wortmann SB. Treatment recommendations for glycogen storage disease type IB- associated neutropenia and neutrophil dysfunction with empagliflozin: Consensus from an international workshop. Molecular genetics and metabolism. 2024 Mar:141(3):108144. doi: 10.1016/j.ymgme.2024.108144. Epub 2024 Jan 17 [PubMed PMID: 38277989]
Level 3 (low-level) evidenceGrünert SC, Derks TGJ, Adrian K, Al-Thihli K, Ballhausen D, Bidiuk J, Bordugo A, Boyer M, Bratkovic D, Brunner-Krainz M, Burlina A, Chakrapani A, Corpeleijn W, Cozens A, Dawson C, Dhamko H, Milosevic MD, Eiroa H, Finezilber Y, Moura de Souza CF, Garcia-Jiménez MC, Gasperini S, Haas D, Häberle J, Halligan R, Fung LH, Hörbe-Blindt A, Horka LM, Huemer M, Uçar SK, Kecman B, Kilavuz S, Kriván G, Lindner M, Lüsebrink N, Makrilakis K, Mei-Kwun Kwok A, Maier EM, Maiorana A, McCandless SE, Mitchell JJ, Mizumoto H, Mundy H, Ochoa C, Pierce K, Fraile PQ, Regier D, Rossi A, Santer R, Schuman HC, Sobieraj P, Spenger J, Spiegel R, Stepien KM, Tal G, Tanšek MZ, Torkar AD, Tchan M, Thyagu S, Schrier Vergano SA, Vucko E, Weinhold N, Zsidegh P, Wortmann SB. Efficacy and safety of empagliflozin in glycogen storage disease type Ib: Data from an international questionnaire. Genetics in medicine : official journal of the American College of Medical Genetics. 2022 Aug:24(8):1781-1788. doi: 10.1016/j.gim.2022.04.001. Epub 2022 May 3 [PubMed PMID: 35503103]
Kaczor M, Greczan M, Kierus K, Ehmke Vel Emczyńska-Seliga E, Ciara E, Piątosa B, Rokicki D, Książyk J, Wesół-Kucharska D. Sodium-glucose cotransporter type 2 channel inhibitor: Breakthrough in the treatment of neutropenia in patients with glycogen storage disease type 1b? JIMD reports. 2022 May:63(3):199-206. doi: 10.1002/jmd2.12278. Epub 2022 Mar 2 [PubMed PMID: 35433171]
Carvalho PM, Silva NJ, Dias PG, Porto JF, Santos LC, Costa JM. Glycogen Storage Disease type 1a - a secondary cause for hyperlipidemia: report of five cases. Journal of diabetes and metabolic disorders. 2013 Jun 6:12(1):25. doi: 10.1186/2251-6581-12-25. Epub 2013 Jun 6 [PubMed PMID: 23738826]
Level 3 (low-level) evidenceGümüş E, Özen H. Glycogen storage diseases: An update. World journal of gastroenterology. 2023 Jul 7:29(25):3932-3963. doi: 10.3748/wjg.v29.i25.3932. Epub [PubMed PMID: 37476587]
Ellison S, Parker H, Bigger B. Advances in therapies for neurological lysosomal storage disorders. Journal of inherited metabolic disease. 2023 Sep:46(5):874-905. doi: 10.1002/jimd.12615. Epub 2023 May 2 [PubMed PMID: 37078180]
Level 3 (low-level) evidenceKishnani PS, Sun B, Koeberl DD. Gene therapy for glycogen storage diseases. Human molecular genetics. 2019 Oct 1:28(R1):R31-R41. doi: 10.1093/hmg/ddz133. Epub [PubMed PMID: 31227835]
Chou JY, Mansfield BC. Gene therapy and genome editing for type I glycogen storage diseases. Frontiers in molecular medicine. 2023:3():1167091. doi: 10.3389/fmmed.2023.1167091. Epub 2023 Mar 31 [PubMed PMID: 39086673]